
Relationship between Brain Metabolic Disorders and Cognitive Impairment: LDL Receptor Defect

 ,
,  , , and
, , and 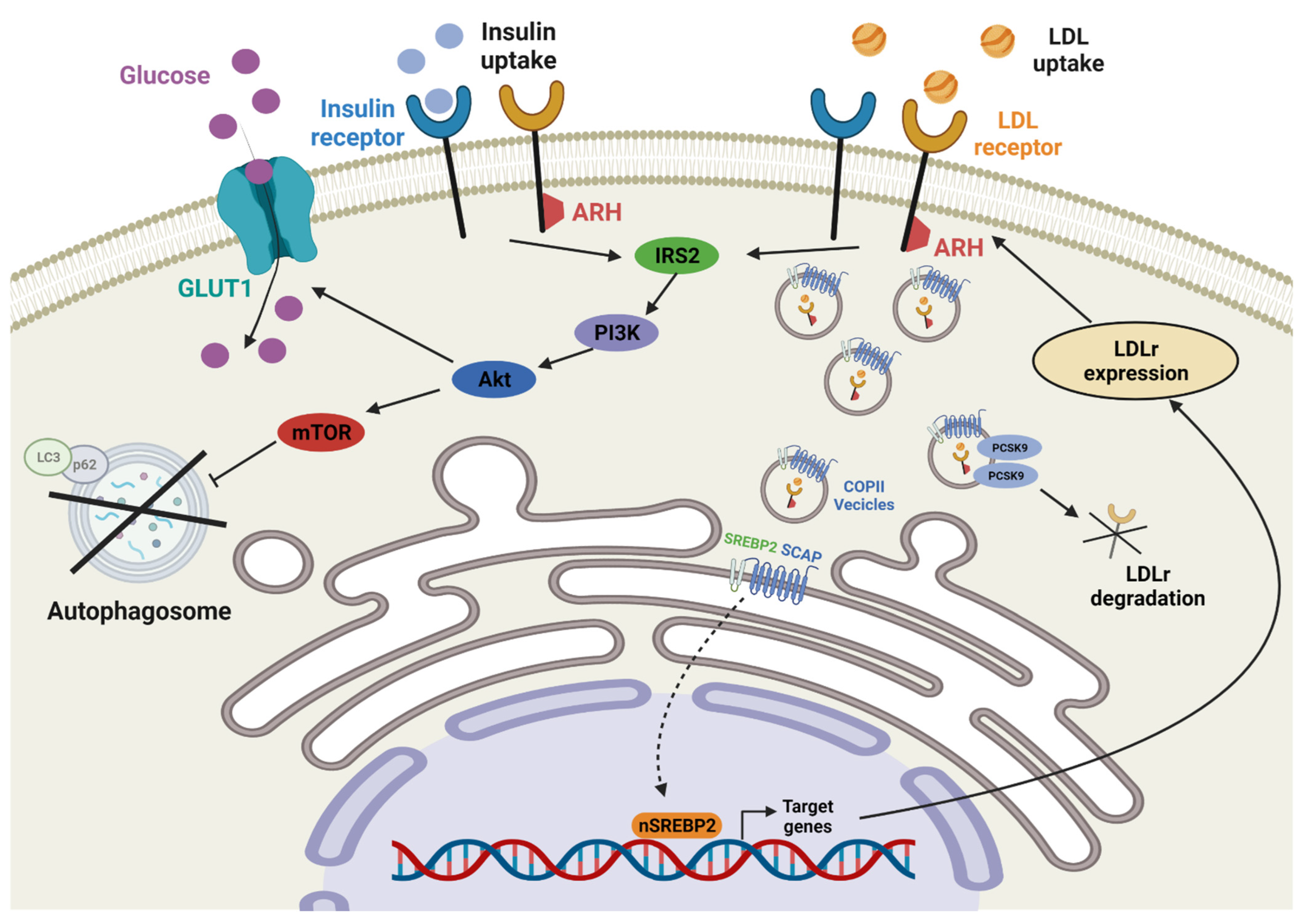{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cholesterol Regulation of LDLr
3. Interaction of LDLr and IR
4. Cholesterol Metabolism in the Brain
5. BBB Breakdown
6. Neuroinflammation
7. Interaction between ER Stress and Mitochondria
8. ApoE
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramakrishnan, G.; Arjuman, A.; Suneja, S.; Das, C.; Chandra, N.C. The association between insulin and low-density lipoprotein receptors. Diabetes Vasc. Dis. Res. 2012, 9, 196–204. [Google Scholar]
- Wade, D.P.; Knight, B.L.; Soutar, A.K. Hormonal regulation of low-density lipoprotein (LDL) receptor activity in human hepatoma Hep G2 cells: Insulin increases LDL receptor activity and diminishes its suppression by exogenous LDL. Eur. J. Biochem. 1988, 174, 213–218. [Google Scholar]
- Goritz, C.; Mauch, D.H.; Pfrieger, F.W. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell. Neurosci. 2005, 29, 190–201. [Google Scholar]
- Pfenninger, K.H. Plasma membrane expansion: A neuron’s Herculean task. Nat. Rev. Neurosci. 2009, 10, 251–261. [Google Scholar]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar]
- Linetti, A.; Fratangeli, A.; Taverna, E.; Valnegri, P.; Francolini, M.; Cappello, V.; Matteoli, M.; Passafaro, M.; Rosa, P. Cholesterol reduction impairs exocytosis of synaptic vesicles. J. Cell Sci. 2010, 123, 595–605. [Google Scholar]
- Liu, Q.; Trotter, J.; Zhang, J.; Peters, M.M.; Cheng, H.; Bao, J.; Han, X.; Weeber, E.J.; Bu, G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 2010, 30, 17068–17078. [Google Scholar]
- Liu, Q.; Zerbinatti, C.V.; Zhang, J.; Hoe, H.-S.; Wang, B.; Cole, S.L.; Herz, J.; Muglia, L.; Bu, G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 2007, 56, 66–78. [Google Scholar]
- Block, R.C.; Dorsey, E.R.; Beck, C.A.; Brenna, J.T.; Shoulson, I. Altered cholesterol and fatty acid metabolism in Huntington disease. J. Clin. Lipidol. 2010, 4, 17–23. [Google Scholar]
- Di Paolo, G.; Kim, T.-W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar]
- Wang, Q.; Yan, J.; Chen, X.; Li, J.; Yang, Y.; Weng, J.; Deng, C.; Yenari, M.A. Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp. Neurol. 2011, 230, 27–34. [Google Scholar]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M.S.; Bhat, N.R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008, 106, 475–485. [Google Scholar]
- Moreira, E.L.G.; de Oliveira, J.; Engel, D.F.; Walz, R.; de Bem, A.F.; Farina, M.; Prediger, R.D.S. Hypercholesterolemia induces short-term spatial memory impairments in mice: Up-regulation of acetylcholinesterase activity as an early and causal event? J. Neural Transm. 2014, 121, 415–426. [Google Scholar]
- Rutkowsky, J.M.; Lee, L.L.; Puchowicz, M.; Golub, M.S.; Befroy, D.E.; Wilson, D.W.; Anderson, S.; Cline, G.; Bini, J.; Borkowski, K.; et al. Reduced cognitive function, increased blood-brain-barrier transport and inflammatory responses, and altered brain metabolites in LDLr−/−and C57BL/6 mice fed a western diet. PLoS ONE 2018, 13, e0191909. [Google Scholar]
- Witte, M.E.; Geurts, J.J.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion 2010, 10, 411–418. [Google Scholar]
- Yadav, R.; Weng, H.-R. EZH2 regulates spinal neuroinflammation in rats with neuropathic pain. Neuroscience 2017, 349, 106–117. [Google Scholar]
- Crisby, M.; Rahman, S.; Sylven, C.; Winblad, B.; Schultzberg, M. Effects of high cholesterol diet on gliosis in apolipoprotein E knockout mice: Implications for Alzheimer’s disease and stroke. Neurosci. Lett. 2004, 369, 87–92. [Google Scholar]
- Montilla, P.; Espejo, I.; Muñoz, M.C.; Bujalance, I.; Muñoz-Castañeda, J.R.; Tunez, I. Protective effect of red wine on oxidative stress and antioxidant enzyme activities in the brain and kidney induced by feeding high cholesterol in rats. Clin. Nutr. 2006, 25, 146–153. [Google Scholar]
- Goldstein, J.L.; Brown, M.S. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J. Biol. Chem. 1974, 249, 5153–5162. [Google Scholar]
- Goldstein, J.L.; Brown, M.S.; Stone, N.J. Genetics of the LDL receptor: Evidence that the mutations affecting binding and internalization are allelic. Cell 1977, 12, 629–641. [Google Scholar]
- Goldstein, J.L.; Dana, S.E.; Brown, M.S. Esterification of low density lipoprotein cholesterol in human fibroblasts and its absence in homozygous familial hypercholesterolemia. Proc. Natl. Acad. Sci. USA 1974, 71, 4288–4292. [Google Scholar]
- Goldstein, J.L.; Brown, M.S. Familial hypercholesterolemia: Identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc. Natl. Acad. Sci. USA 1973, 70, 2804–2808. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. Familial hypercholesterolemia: Defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc. Natl. Acad. Sci. USA 1974, 71, 788–792. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. Expression of the familial hypercholesterolemia gene in heterozygotes: Mechanism for a dominant disorder in man. Science 1974, 185, 61–63. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar]
- Horton, J.D.; Shah, N.A.; Warrington, J.A.; Anderson, N.N.; Park, S.W.; Brown, M.S.; Goldstein, J.L. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. USA 2003, 100, 12027–12032. [Google Scholar]
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008, 8, 512–521. [Google Scholar]
- Yang, H.X.; Zhang, M.; Long, S.Y.; Tuo, Q.H.; Tian, Y.; Chen, J.X.; Zhang, C.P.; Liao, D.F. Cholesterol in LDL receptor recycling and degradation. Clin. Chim. Acta 2020, 500, 81–86. [Google Scholar]
- Sun, L.P.; Li, L.; Goldstein, J.L.; Brown, M.S. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 2005, 280, 26483–26490. [Google Scholar]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar]
- Yabe, D.; Brown, M.S.; Goldstein, J.L. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 12753–12758. [Google Scholar]
- Garcia, C.K.; Wilund, K.; Arca, M.; Zuliani, G.; Fellin, R.; Maioli, M.; Calandra, S.; Bertolini, S.; Cossu, F.; Grishin, N.; et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 2001, 292, 1394–1398. [Google Scholar]
- He, G.; Gupta, S.; Yi, M.; Michaely, P.; Hobbs, H.H.; Cohen, J.C. ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J. Biol. Chem. 2002, 277, 44044–44049. [Google Scholar]
- Michaely, P.; Li, W.P.; Anderson, R.G.; Cohen, J.C.; Hobbs, H.H. The modular adaptor protein ARH is required for low density lipoprotein (LDL) binding and internalization but not for LDL receptor clustering in coated pits. J. Biol. Chem. 2004, 279, 34023–34031. [Google Scholar]
- Zhao, Z.; Pompey, S.; Dong, H.; Weng, J.; Garuti, R.; Michaely, P. S-nitrosylation of ARH is required for LDL uptake by the LDL receptor. J. Lipid Res. 2013, 54, 1550–1559. [Google Scholar]
- Tao, W.; Moore, R.; Meng, Y.; Smith, E.R.; Xu, X.X. Endocytic adaptors Arh and Dab2 control homeostasis of circulatory cholesterol. J. Lipid Res. 2016, 57, 809–817. [Google Scholar]
- Park, S.W.; Moon, Y.A.; Horton, J.D. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 2004, 279, 50630–50638. [Google Scholar]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar]
- Goldstein, J.L.; Brown, M.S. The low-density lipoprotein pathway and its relation to atherosclerosis. Annu. Rev. Biochem. 1977, 46, 897–930. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar]
- Jeon, H.; Blacklow, S.C. Structure and physiologic function of the low-density lipoprotein receptor. Annu. Rev. Biochem. 2005, 74, 535–562. [Google Scholar]
- Go, G.W.; Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J. Biol. Med. 2012, 85, 19–28. [Google Scholar]
- Anderson, R.G.; Brown, M.S.; Goldstein, J.L. Role of the coated endocytic vesicle in the uptake of receptor-bound low density lipoprotein in human fibroblasts. Cell 1977, 10, 351–364. [Google Scholar]
- De Meyts, P. Insulin and its receptor: Structure, function and evolution. Bioessays 2004, 26, 1351–1362. [Google Scholar]
- De Meyts, P. The insulin receptor: A prototype for dimeric, allosteric membrane receptors? Trends Biochem. Sci. 2008, 33, 376–384. [Google Scholar]
- Youngren, J.F. Regulation of insulin receptor function. Cell Mol. Life Sci. 2007, 64, 873–891. [Google Scholar]
- Steiner, G. The dyslipoproteinemias of diabetes. Atherosclerosis 1994, 110, S27–S33. [Google Scholar]
- Ilonen, J.; Lempainen, J.; Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 635–650. [Google Scholar]
- Kahn, S.E.; Cooper, M.E.; del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar]
- Ozougwu, J.; Obimba, K.; Belonwu, C.; Unakalamba, C. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J. Physiol. Pathophysiol. 2013, 4, 46–57. [Google Scholar]
- Guerre-Millo, M.; Lavau, M.; Horne, J.S.; Wardzala, L.J. Proposed mechanism for increased insulin-mediated glucose transport in adipose cells from young, obese Zucker rats. Large intracellular pool of glucose transporters. J. Biol. Chem. 1985, 260, 2197–2201. [Google Scholar]
- Ill, C.; Lepine, J.; Gospodarowicz, D. Permissive effect of insulin on the adenosine 3′,5′-monophosphate-dependent up-regulation of low density lipoprotein receptors and the stimulation of steroid release in bovine adrenal cortical cells. Endocrinology 1984, 114, 767–775. [Google Scholar]
- Chandra, N.C. A comprehensive account of insulin and LDL receptor activity over the years: A highlight on their signaling and functional role. J. Biochem. Mol. Toxicol. 2021, 35, e22840. [Google Scholar]
- Zhu, L.; Wu, G.; Yang, X.; Jia, X.; Li, J.; Bai, X.; Li, W.; Zhao, Y.; Li, Y.; Cheng, W.; et al. Low density lipoprotein mimics insulin action on autophagy and glucose uptake in endothelial cells. Sci. Rep. 2019, 9, 3020. [Google Scholar]
- Tang, F.; Yang, T.L. MicroRNA-126 alleviates endothelial cells injury in atherosclerosis by restoring autophagic flux via inhibiting of PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 495, 1482–1489. [Google Scholar]
- Obici, S.; Feng, Z.; Karkanias, G.; Baskin, D.G.; Rossetti, L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 2002, 5, 566–572. [Google Scholar]
- Liu, T.; Li, Y.; Yang, B.; Wang, H.; Lu, C.; Chang, A.K.; Huang, X.; Zhang, X.; Lu, Z.; Lu, X.; et al. Suppression of neuronal cholesterol biosynthesis impairs brain functions through insulin-like growth factor I-Akt signaling. Int. J. Biol. Sci. 2021, 17, 3702–3716. [Google Scholar]
- Blokland, A.; Jolles, J. Spatial learning deficit and reduced hippocampal ChAT activity in rats after an ICV injection of streptozotocin. Pharmacol. Biochem. Behav. 1993, 44, 491–494. [Google Scholar]
- Kinoshita, J.; Fagan, A.; Ewbank, D.; Marlatt, M.; Heyn, P.; de la Monte, S.; Lombardo, N.E. Alzheimer Research Forum live discussion: Insulin resistance: A common axis linking Alzheimer’s, depression, and metabolism? J. Alzheimers Dis. 2006, 9, 89–93. [Google Scholar]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W., Jr.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimers Dis. 2005, 7, 103–117. [Google Scholar]
- de la Monte, S.M.; Tong, M.; Lester-Coll, N.; Plater, M., Jr.; Wands, J.R. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: Relevance to Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 89–109. [Google Scholar]
- Lester-Coll, N.; Rivera, E.J.; Soscia, S.J.; Doiron, K.; Wands, J.R.; de la Monte, S.M. Intracerebral streptozotocin model of type 3 diabetes: Relevance to sporadic Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 13–33. [Google Scholar]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High Cholesterol/Low Cholesterol: Effects in Biological Membranes: A Review. Cell Biochem. Biophys. 2017, 7, 369–385. [Google Scholar]
- Gliozzi, M.; Musolino, V.; Bosco, F.; Scicchitano, M.; Scarano, F.; Nucera, S.; Zito, M.C.; Ruga, S.; Carresi, C.; Macrì, R.; et al. Cholesterol homeostasis: Researching a dialogue between the brain and peripheral tissues. Pharmacol. Res. 2021, 163, 105215. [Google Scholar]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar]
- Howe, V.; Sharpe, L.J.; Alexopoulos, S.J.; Kunze, S.V.; Chua, N.K.; Li, D.; Brown, A.J. Cholesterol homeostasis: How do cells sense sterol excess? Chem. Phys. Lipids 2016, 199, 170–178. [Google Scholar]
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar]
- Chen, Y.Q.; Troutt, J.S.; Konrad, R.J. PCSK9 is present in human cerebrospinal fluid and is maintained at remarkably constant concentrations throughout the course of the day. Lipids 2014, 49, 445–455. [Google Scholar]
- Zimetti, F.; Caffarra, P.; Ronda, N.; Favari, E.; Adorni, M.P.; Zanotti, I.; Bernini, F.; Barocco, F.; Spallazzi, M.; Galimberti, D.; et al. Increased PCSK9 Cerebrospinal Fluid Concentrations in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 315–320. [Google Scholar]
- Adorni, M.P.; Ruscica, M.; Ferri, N.; Bernini, F.; Zimetti, F. Proprotein Convertase Subtilisin/Kexin Type 9, Brain Cholesterol Homeostasis and Potential Implication for Alzheimer’s Disease. Front Aging Neurosci. 2019, 11, 120. [Google Scholar]
- de Oliveira, J.; Engel, D.F.; de Paula, G.C.; Santos, D.B.D.; Lopes, J.B.; Farina, M.; Moreira, E.L.G.; de Bem, A.F. High Cholesterol Diet Exacerbates Blood-Brain Barrier Disruption in LDLr−/− Mice: Impact on Cognitive Function. J. Alzheimers Dis. 2020, 78, 97–115. [Google Scholar]
- Tung, H.; Lin, H.J.; Chen, P.L.; Lu, T.J.; Jhan, P.P.; Chen, J.P.; Chen, Y.M.; Wu, C.C.; Lin, Y.Y.; Hsiao, T.-H. Characterization of familial hypercholesterolemia in Taiwanese ischemic stroke patients. Aging 2021, 13, 19339–19351. [Google Scholar]
- Khera, A.V.; Won, H.H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll Cardiol. 2016, 67, 2578–2589. [Google Scholar]
- Mahdieh, N.; Heshmatzad, K.; Rabbani, B. A systematic review of LDLR, PCSK9, and APOB variants in Asia. Atherosclerosis 2020, 305, 50–57. [Google Scholar]
- Meshkov, A.; Ershova, A.; Kiseleva, A.; Zotova, E.; Sotnikova, E.; Petukhova, A.; Zharikova, A.; Malyshev, P.; Rozhkova, T.; Limonova, A.; et al. The LDLR, APOB, and PCSK9 Variants of Index Patients with Familial Hypercholesterolemia in Russia. Genes 2021, 12, 66. [Google Scholar]
- Wolburg, H.; Lippoldt, A. Tight junctions of the blood-brain barrier: Development, composition and regulation. Vascul Pharmacol. 2002, 38, 323–337. [Google Scholar]
- Li, W.; Chen, Z.; Chin, I.; Chen, Z.; Dai, H. The Role of VE-cadherin in Blood-brain Barrier Integrity Under Central Nervous System Pathological Conditions. Curr. Neuropharmacol. 2018, 16, 1375–1384. [Google Scholar]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar]
- Tucsek, Z.; Toth, P.; Tarantini, S.; Sosnowska, D.; Gautam, T.; Warrington, J.P.; Giles, C.B.; Wren, J.D.; Koller, A.; Ballabh, P.; et al. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J. Gerontol. Biol. Sci. Med. Sci. 2014, 69, 1339–1352. [Google Scholar]
- Deng, J.; Zhao, F.; Yu, X.; Zhao, Y.; Li, D.; Shi, H.; Sun, Y. Expression of aquaporin 4 and breakdown of the blood-brain barrier after hypoglycemia-induced brain edema in rats. PLoS ONE 2014, 9, e107022. [Google Scholar]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar]
- Karpouzas, G.A.; Ormseth, S.R.; Ronda, N.; Hernandez, E.; Budoff, M.J. Lipoprotein oxidation may underlie the paradoxical association of low cholesterol with coronary atherosclerotic risk in rheumatoid arthritis. J. Autoimmun. 2022, 129, 102815. [Google Scholar]
- Saad, A.F.; Virella, G.; Chassereau, C.; Boackle, R.J.; Lopes-Virella, M.F. OxLDL immune complexes activate complement and induce cytokine production by MonoMac 6 cells and human macrophages. J. Lipid Res. 2006, 47, 1975–1983. [Google Scholar]
- Morris, G.; Berk, M.; Walder, K.; O’Neil, A.; Maes, M.; Puri, B.K. The lipid paradox in neuroprogressive disorders: Causes and consequences. Neurosci. Biobehav. Rev. 2021, 128, 35–57. [Google Scholar]
- Saher, G.; Stumpf, S.K. Cholesterol in myelin biogenesis and hypomyelinating disorders. Biochim. Biophys. Acta 2015, 1851, 1083–1094. [Google Scholar]
- Sun, R.; Peng, M.; Xu, P.; Huang, F.; Xie, Y.; Li, J.; Hong, Y.; Guo, H.; Liu, Q.; Zhu, W. Low-density lipoprotein receptor (LDLR) regulates NLRP3-mediated neuronal pyroptosis following cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2020, 17, 330. [Google Scholar]
- Lee, D.H.; Lee, J.Y.; Hong, D.Y.; Lee, E.C.; Park, S.W.; Lee, M.R.; Oh, J.S. Neuroinflammation in Post-Traumatic Stress Disorder. Biomedicines 2022, 10, 953. [Google Scholar]
- Bzowska, M.; Nogieć, A.; Skrzeczyńska-Moncznik, J.; Mickowska, B.; Guzik, K.; Pryjma, J. Oxidized LDLs inhibit TLR-induced IL-10 production by monocytes: A new aspect of pathogen-accelerated atherosclerosis. Inflammation 2012, 35, 1567–1584. [Google Scholar]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 2016, 13, 148–159. [Google Scholar]
- Barrington, J.; Lemarchand, E.; Allan, S.M. A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol. 2017, 27, 205–212. [Google Scholar]
- Malik, A.; Kanneganti, T.D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar]
- Zhang, D.; Qian, J.; Zhang, P.; Li, H.; Shen, H.; Li, X.; Chen, G. Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J. Neurosci. Res. 2019, 97, 645–660. [Google Scholar]
- Wang, Q.; Wu, J.; Zeng, Y.; Chen, K.; Wang, C.; Yang, S.; Sun, N.; Chen, H.; Duan, K.; Zeng, G. Pyroptosis: A pro-inflammatory type of cell death in cardiovascular disease. Clin. Chim. Acta 2020, 510, 62–72. [Google Scholar]
- Opoku, E.; Traughber, C.A.; Zhang, D.; Iacano, A.J.; Khan, M.; Han, J.; Smith, J.D.; Gulshan, K. Gasdermin D Mediates Inflammation-Induced Defects in Reverse Cholesterol Transport and Promotes Atherosclerosis. Front Cell Dev. Biol. 2021, 9, 715211. [Google Scholar]
- Tumurkhuu, G.; Shimada, K.; Dagvadorj, J.; Crother, T.R.; Zhang, W.; Luthringer, D.; Gottlieb, R.A.; Chen, S.; Arditi, M. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ. Res. 2016, 119, e76–e90. [Google Scholar]
- Levels, J.H.; Marquart, J.A.; Abraham, P.R.; van den Ende, A.E.; Molhuizen, H.O.; van Deventer, S.J.; Meijers, J.C. Lipopolysaccharide is transferred from high-density to low-density lipoproteins by lipopolysaccharide-binding protein and phospholipid transfer protein. Infect Immun. 2005, 73, 2321–2326. [Google Scholar]
- Walley, K.R.; Thain, K.R.; Russell, J.A.; Reilly, M.P.; Meyer, N.J.; Ferguson, J.F.; Christie, J.D.; Nakada, T.A.; Fjell, C.D.; Thair, S.A.; et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci. Transl. Med. 2014, 6, 258ra143. [Google Scholar]
- Tang, Z.H.; Li, T.H.; Peng, J.; Zheng, J.; Li, T.T.; Liu, L.S.; Jiang, Z.S.; Zheng, X.L. PCSK9: A novel inflammation modulator in atherosclerosis? J. Cell Physiol. 2019, 234, 2345–2355. [Google Scholar]
- Chen, H.; Chan, D.C. Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell Biol. 2006, 18, 453–459. [Google Scholar]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke. 2018, 13, 612–632. [Google Scholar]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar]
- Kizhakkedath, P.; John, A.; Al-Sawafi, B.K.; Al-Gazali, L.; Ali, B.R. Endoplasmic reticulum quality control of LDLR variants associated with familial hypercholesterolemia. FEBS Open Bio. 2019, 9, 1994–2005. [Google Scholar]
- Sørensen, S.; Ranheim, T.; Bakken, K.S.; Leren, T.P.; Kulseth, M.A. Retention of mutant low density lipoprotein receptor in endoplasmic reticulum (ER) leads to ER stress. J. Biol. Chem. 2006, 281, 468–476. [Google Scholar]
- Oommen, D.; Kizhakkedath, P.; Jawabri, A.A.; Varghese, D.S.; Ali, B.R. Proteostasis Regulation in the Endoplasmic Reticulum: An Emerging Theme in the Molecular Pathology and Therapeutic Management of Familial Hypercholesterolemia. Front Genet. 2020, 11, 570355. [Google Scholar]
- El-Bachá, R.S.; De-Lima-Filho, J.L.; Guedes, R.C. Dietary Antioxidant Deficiency Facilitates Cortical Spreading Depression Induced by Photoactivated Riboflavin. Nutr. Neurosci. 1998, 1, 205–212. [Google Scholar]
- Mandel, S.; Grünblatt, E.; Riederer, P.; Gerlach, M.; Levites, Y.; Youdim, M.B. Neuroprotective strategies in Parkinson’s disease: An update on progress. CNS Drugs 2003, 17, 729–762. [Google Scholar]
- Yu, Y.C.; Kuo, C.L.; Cheng, W.L.; Liu, C.S.; Hsieh, M. Decreased antioxidant enzyme activity and increased mitochondrial DNA damage in cellular models of Machado-Joseph disease. J. Neurosci. Res. 2009, 87, 1884–1891. [Google Scholar]
- Zeya, B.; Arjuman, A.; Chandra, N.C. Lectin-like Oxidized Low-Density Lipoprotein (LDL) Receptor (LOX-1): A Chameleon Receptor for Oxidized LDL. Biochemistry 2016, 55, 4437–4444. [Google Scholar]
- Kattoor, A.J.; Kanuri, S.H.; Mehta, J.L. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 2019, 26, 1693–1700. [Google Scholar]
- Yu, S.; Zhang, L.; Liu, C.; Yang, J.; Zhang, J.; Huang, L. PACS2 is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca(2+) elevation. Exp. Cell Res. 2019, 379, 191–202. [Google Scholar]
- Nègre-Salvayre, A.; Augé, N.; Camaré, C.; Bacchetti, T.; Ferretti, G.; Salvayre, R. Dual signaling evoked by oxidized LDLs in vascular cells. Free Radic. Biol. Med. 2017, 106, 118–133. [Google Scholar]
- Lubrano, V.; Balzan, S. LOX-1 and ROS, inseparable factors in the process of endothelial damage. Free Radic Res. 2014, 48, 841–848. [Google Scholar]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calò, L.A. The role of oxidized low-density lipoproteins in atherosclerosis: The myths and the facts. Mediat. Inflamm. 2013, 2013, 714653. [Google Scholar]
- Zhou, Y.D.; Cao, X.Q.; Liu, Z.H.; Cao, Y.J.; Liu, C.F.; Zhang, Y.L.; Xie, Y. Rapamycin Inhibits Oxidized Low Density Lipoprotein Uptake in Human Umbilical Vein Endothelial Cells via mTOR/NF-κB/LOX-1 Pathway. PLoS ONE 2016, 11, e0146777. [Google Scholar]
- Xu, S.; Ogura, S.; Chen, J.; Little, P.J.; Moss, J.; Liu, P. LOX-1 in atherosclerosis: Biological functions and pharmacological modifiers. Cell Mol. Life Sci. 2013, 70, 2859–2872. [Google Scholar]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler Rep. 2017, 19, 42. [Google Scholar]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J., Jr.; Davies, P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar]
- Yang, S.; Wu, M.; Li, X.; Zhao, R.; Zhao, Y.; Liu, L.; Wang, S. Role of Endoplasmic Reticulum Stress in Atherosclerosis and Its Potential as a Therapeutic Target. Oxid Med. Cell Longev. 2020, 2020, 9270107. [Google Scholar]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct mPTP activation mechanisms in ischaemia-reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529 Pt 1, 57–68. [Google Scholar]
- Martínez-Klimova, E.; Aparicio-Trejo, O.E.; Gómez-Sierra, T.; Jiménez-Uribe, A.P.; Bellido, B.; Pedraza-Chaverri, J. Mitochondrial dysfunction and endoplasmic reticulum stress in the promotion of fibrosis in obstructive nephropathy induced by unilateral ureteral obstruction. Biofactors 2020, 46, 716–733. [Google Scholar]
- Borutaite, V.; Toleikis, A.; Brown, G.C. In the eye of the storm: Mitochondrial damage during heart and brain ischaemia. FEBS J. 2013, 280, 4999–5014. [Google Scholar]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123 Pt 19 Pt 19, 3209–3214. [Google Scholar]
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar]
- de Oliveira, J.; Hort, M.A.; Moreira, E.L.; Glaser, V.; Ribeiro-do-Valle, R.M.; Prediger, R.D.; Farina, M.; Latini, A.; de Bem, A.F. Positive correlation between elevated plasma cholesterol levels and cognitive impairments in LDL receptor knockout mice: Relevance of cortico-cerebral mitochondrial dysfunction and oxidative stress. Neuroscience 2011, 197, 99–106. [Google Scholar]
- Han, Z.; Wang, Y.; Li, J. Effects of Atorvastatin Combined with Nano-Selenium on Blood Lipids and Oxidative Stress in Atherosclerotic Rats. J. Nanosci. Nanotechnol. 2021, 21, 1331–1337. [Google Scholar]
- Moreira, E.L.; de Oliveira, J.; Nunes, J.C.; Santos, D.B.; Nunes, F.C.; Vieira, D.S.; Ribeiro-do-Valle, R.M.; Pamplona, F.A.; de Bem, A.F.; Farina, M.; et al. Age-related cognitive decline in hypercholesterolemic LDL receptor knockout mice (LDLr−/−): Evidence of antioxidant imbalance and increased acetylcholinesterase activity in the prefrontal cortex. J. Alzheimers Dis. 2012, 32, 495–511. [Google Scholar]
- Casula, M.; Catapano, A.L.; Bernardi, L.R.; Visconti, M.; Aronica, A. Detection of familial hypercholesterolemia in patients from a general practice database. Atheroscler Suppl. 2017, 29, 25–30. [Google Scholar]
- El Shamieh, S.; Costanian, C.; Kassir, R.; Visvkis-Siest, S.; Bissar-Tadmouri, N. APOE genotypes in Lebanon: Distribution and association with hypercholesterolemia and Alzheimer’s disease. Per Med. 2019, 16, 15–23. [Google Scholar]
- Shi, Y.; Andhey, P.S.; Ising, C.; Wang, K.; Snipes, L.L.; Boyer, K.; Lawson, S.; Yamada, K.; Qin, W.; Manis, M.; et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron 2021, 109, 2413–2426.e7. [Google Scholar]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar]
- Rhea, E.M.; Banks, W.A. Interactions of Lipids, Lipoproteins, and Apolipoproteins with the Blood-Brain Barrier. Pharm. Res. 2021, 38, 1469–1475. [Google Scholar]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar]
- Wisniewski, T.; Frangione, B. Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 1992, 135, 235–238. [Google Scholar]
- Jordan, B.D.; Relkin, N.R.; Ravdin, L.D.; Jacobs, A.R.; Bennett, A.; Gandy, S. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA 1997, 278, 136–140. [Google Scholar]
- Mayeux, R.; Ottman, R.; Maestre, G.; Ngai, C.; Tang, M.X.; Ginsberg, H.; Chun, M.; Tycko, B.; Shelanski, M. Synergistic effects of traumatic head injury and apolipoprotein-epsilon 4 in patients with Alzheimer’s disease. Neurology 1995, 45 Pt 1, 555–557. [Google Scholar]
- Mannix, R.C.; Zhang, J.; Park, J.; Zhang, X.; Bilal, K.; Walker, K.; Tanzi, R.E.; Tesco, G.; Whalen, M.J. Age-dependent effect of apolipoprotein E4 on functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 2011, 31, 351–361. [Google Scholar]
- Golde, T.E.; Schneider, L.S.; Koo, E.H. Anti-aβ therapeutics in Alzheimer’s disease: The need for a paradigm shift. Neuron 2011, 69, 203–213. [Google Scholar]
- Benilova, I.; Karran, E.; de Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Basak, J.M.; Verghese, P.B.; Yoon, H.; Kim, J.; Holtzman, D.M. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J. Biol. Chem. 2012, 287, 13959–13971. [Google Scholar]
- Castellano, J.M.; Deane, R.; Gottesdiener, A.J.; Verghese, P.B.; Stewart, F.R.; West, T.; Paoletti, A.C.; Kasper, T.R.; DeMattos, R.B.; Zlokovic, B.V.; et al. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc. Natl. Acad. Sci. USA 2012, 109, 15502–15507. [Google Scholar]
- de Oliveira, J.; Moreira, E.L.; Santos, D.B.d.; Piermartiri, T.C.; Dutra, R.C.; Pinton, S.; Tasca, C.I.; Farina, M.; Prediger, R.D.; de Bem, A.F. Increased susceptibility to amyloid-β-induced neurotoxicity in mice lacking the low-density lipoprotein receptor. J. Alzheimers Dis. 2014, 41, 43–60. [Google Scholar]
- Zhao, Y.; Yang, Y.; Xing, R.; Cui, X.; Xiao, Y.; Xie, L.; You, P.; Wang, T.; Zeng, L.; Peng, W.; et al. Hyperlipidemia induces typical atherosclerosis development in Ldlr and Apoe deficient rats. Atherosclerosis 2018, 271, 26–35. [Google Scholar]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar]
- Mulder, M.; Jansen, P.J.; Janssen, B.J.; van de Berg, W.D.; van der Boom, H.; Havekes, L.M.; de Kloet, R.E.; Ramaekers, F.C.; Blokland, A. Low-density lipoprotein receptor-knockout mice display impaired spatial memory associated with a decreased synaptic density in the hippocampus. Neurobiol. Dis. 2004, 16, 212–219. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, D.-Y.; Lee, D.-H.; Lee, J.-Y.; Lee, E.-C.; Park, S.-W.; Lee, M.-R.; Oh, J.-S. Relationship between Brain Metabolic Disorders and Cognitive Impairment: LDL Receptor Defect. Int. J. Mol. Sci. 2022, 23, 8384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158384
Hong D-Y, Lee D-H, Lee J-Y, Lee E-C, Park S-W, Lee M-R, Oh J-S. Relationship between Brain Metabolic Disorders and Cognitive Impairment: LDL Receptor Defect. International Journal of Molecular Sciences. 2022; 23(15):8384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158384
Chicago/Turabian StyleHong, Dong-Yong, Dong-Hun Lee, Ji-Young Lee, Eun-Chae Lee, Sang-Won Park, Man-Ryul Lee, and Jae-Sang Oh. 2022. "Relationship between Brain Metabolic Disorders and Cognitive Impairment: LDL Receptor Defect" International Journal of Molecular Sciences 23, no. 15: 8384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158384