
Protein Profiling of a Cellular Model of NAFLD by Advanced Bioanalytical Approaches

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results and Discussion
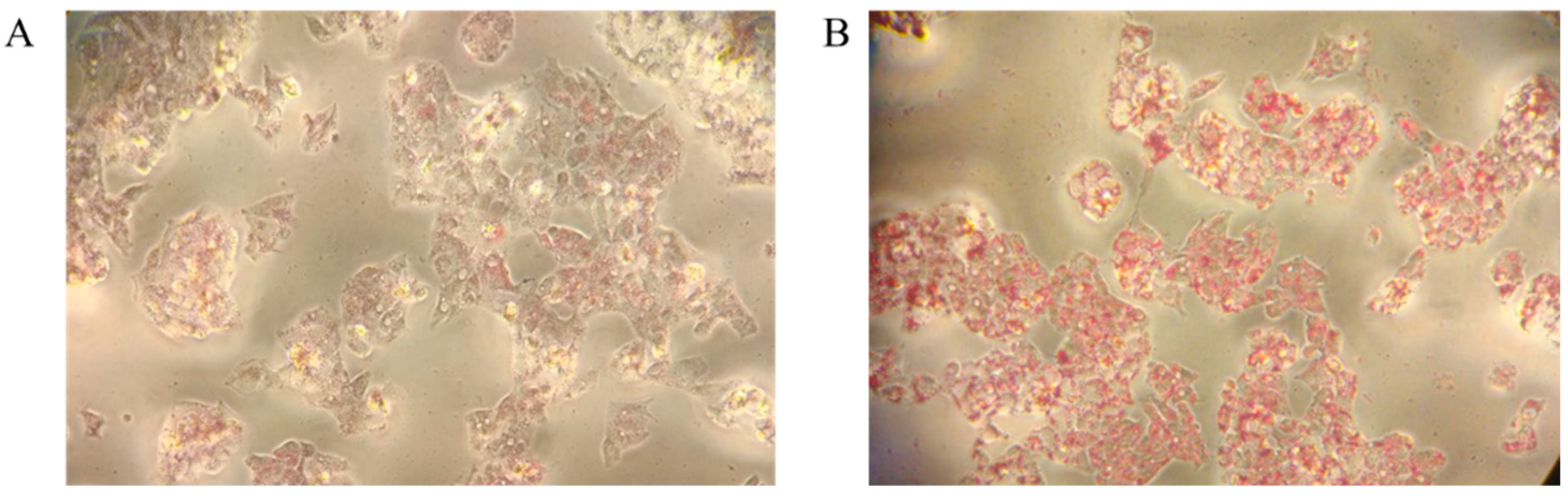
2.1. Oleic-Acid-Induced NAFLD in HepG2 Cells: Validation of the Model
2.2. Description of the NAFLD Proteome by Quantitative Approaches: LFQ, SILAC, and TMT
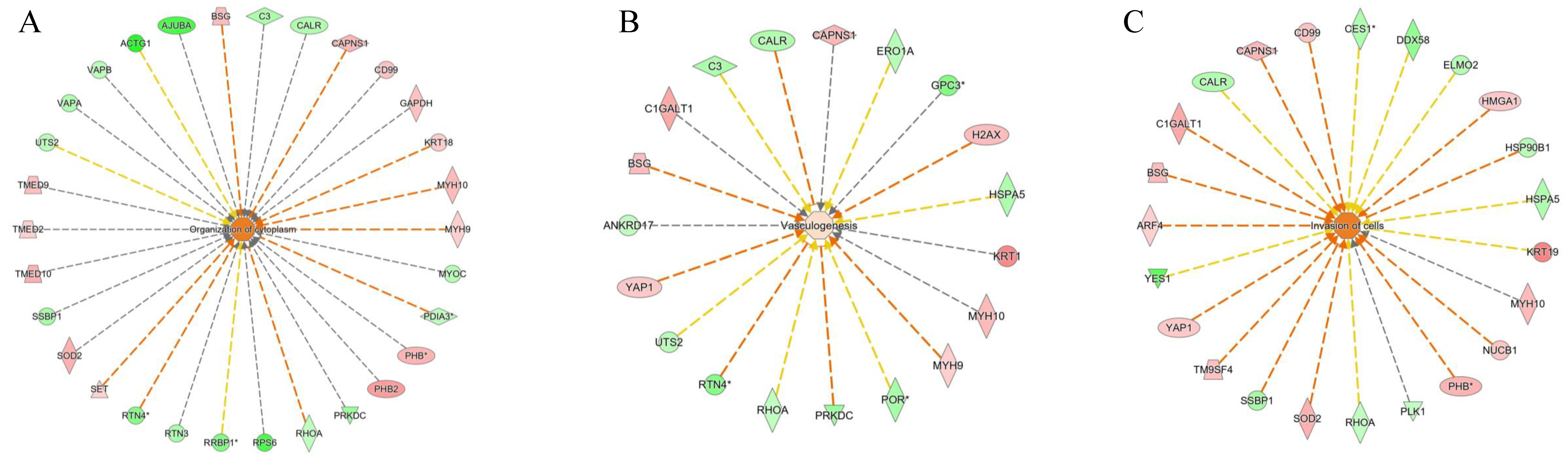
2.3. Description of the NAFLD Proteome by Network Analyses Using Quantitative Data
3. Materials and Methods
3.1. Reagents
3.2. Cell Culture
3.3. Induction of Steatosis
3.4. Oil Red O Staining and Intracellular Lipid Content Assay
3.5. Western Blots and Densitometric Analyses
3.6. Protein Extraction, In-Solution Trypsin Digestion, and TMT Labeling
3.7. Protein Extraction and In-Solution Trypsin Digestion of SILAC and Label-Free Samples
3.8. High-Resolution Quantitative Liquid Chromatography with Tandem Mass Spectrometry (LC–MS/MS) Analysis
3.9. Data Analysis: Protein Quantification and Statistics
3.10. Network Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Slaughter, V.L.; Rumsey, J.W.; Boone, R.; Malik, D.; Cai, Y.Q.; Sriram, N.N.; Long, C.J.; McAleer, C.W.; Lambert, S.; Shuler, M.L.; et al. Validation of an adipose-liver human-on-a-chip model of nafld for preclinical therapeutic efficacy evaluation. Sci. Rep. 2021, 11, 13159. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, H.; Omidian, K.; Bandy, B. Dietary polyphenols protect against oleic acid-induced steatosis in an in vitro model of nafld by modulating lipid metabolism and improving mitochondrial function. Nutrients 2019, 11, 541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, S.D.; Wang, P.; Guo, N.; Wang, W.; Yao, L.P.; Yang, Q.; Efferth, T.; Jiao, J.; Fu, Y.J. Pinolenic acid ameliorates oleic acid-induced lipogenesis and oxidative stress via ampk/sirt1 signaling pathway in hepg2 cells. Eur. J. Pharmacol. 2019, 861, 172618. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.Q.; Liu, Y.Y.; Wu, Y.F.; Chen, Y. Dihydrocurcumin ameliorates the lipid accumulation, oxidative stress and insulin resistance in oleic acid-induced l02 and hepg2 cells. Biomed. Pharmacother. 2018, 103, 1327–1336. [Google Scholar] [CrossRef]
- Zhou, Y.P.; Wu, R.; Shen, W.; Yu, H.H.; Yu, S.J. Comparison of effects of oleic acid and palmitic acid on lipid deposition and mtor/s6k1/srebp-1c pathway in hepg2 cells. Zhonghua Gan Zang Bing Za Zhi 2018, 26, 451–456. [Google Scholar]
- Moravcova, A.; Cervinkova, Z.; Kucera, O.; Mezera, V.; Rychtrmoc, D.; Lotkova, H. The effect of oleic and palmitic acid on induction of steatosis and cytotoxicity on rat hepatocytes in primary culture. Physiol. Res. 2015, 64, S627–S636. [Google Scholar] [CrossRef]
- Araya, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Orellana, M.; Pettinelli, P.; Poniachik, J. Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatiosis in patients with non-alcoholic fatty liver disease. Clin. Sci. 2004, 106, 635–643. [Google Scholar] [CrossRef]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef]
- Zhong, W.X.; Fan, B.; Cong, H.Y.; Wang, T.Y.; Gu, J.Q. Oleic acid-induced perilipin 5 expression and lipid droplets formation are regulated by the pi3k/ppar alpha pathway in hepg2 cells. Appl. Physiol. Nutr. Metab. 2019, 44, 840–848. [Google Scholar] [CrossRef]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. Nlrp3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Jiang, W.T. The effects of rki-1447 in a mouse model of nonalcoholic fatty liver disease induced by a high-fat diet and in hepg2 human hepatocellular carcinoma cells treated with oleic acid. Med. Sci. Monit. 2020, 26, e919220. [Google Scholar] [CrossRef] [PubMed]
- Megger, D.A.; Bracht, T.; Meyer, H.E.; Sitek, B. Label-free quantification in clinical proteomics. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Wei, S.S.; Ji, Y.L.; Guo, X.J.; Yang, F.Q. Quantitative proteomics using silac: Principles, applications, and developments. Proteomics 2015, 15, 3175–3192. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by ms/ms. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Megger, D.A.; Pott, L.L.; Ahrens, M.; Padden, J.; Bracht, T.; Kuhlmann, K.; Eisenacher, M.; Meyer, H.E.; Sitek, B. Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 967–976. [Google Scholar] [CrossRef]
- Cui, W.; Chen, S.L.; Hu, K.Q. Quantification and mechanisms of oleic acid-induced steatosis in hepg2 cells. Am. J. Transl. Res. 2010, 2, 95–104. [Google Scholar]
- Yan, D.; Dou, Q.L.; Wang, Z.; Wei, Y.Y. Establishment of a hepatocyte steatosis model using chang liver cells. Genet. Mol. Res. 2015, 14, 15224–15232. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Itabe, H.; Kinoshita, T.; Homma, K.J.; Onoduka, J.; Mori, M.; Yamaguchi, S.; Makita, M.; Higashi, Y.; Yamashita, A.; et al. Involvement of acsl in local synthesis of neutral lipids in cytoplasmic lipid droplets in human hepatocyte huh7. J. Lipid Res. 2007, 48, 1280–1292. [Google Scholar] [CrossRef]
- Niture, S.; Gyamfi, M.A.; Kedir, H.; Arthur, E.; Ressom, H.; Deep, G.; Kumar, D. Serotonin induced hepatic steatosis is associated with modulation of autophagy and notch signaling pathway. Cell Commun. Signal. 2018, 16, 78. [Google Scholar] [CrossRef]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Kok, C.Y.Y.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.W.; Zhou, Z.X.; Lim, C.W.; Kim, B. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Moreto, F.; Ferron, A.J.T.; Francisqueti-Ferron, F.V.; D’Amato, A.; Garcia, J.L.; Costa, M.R.; Silva, C.; Altomare, A.; Correa, C.R.; Aldini, G.; et al. Differentially expressed proteins obtained by label-free quantitative proteomic analysis reveal affected biological processes and functions in western diet-induced steatohepatitis. Biochem. Mol. Toxicol. 2021, 35, e22751. [Google Scholar] [CrossRef]
- Arteel, G.E.; Naba, A. The liver matrisome—Looking beyond collagens. JHEP Rep. 2020, 2, 100115. [Google Scholar] [CrossRef]
- Pereira, R.M.; dos Santos, R.A.S.; Dias, F.L.D.; Teixeira, M.M.; Silva, A. Renin-angiotensin system in the pathogenesis of liver fibrosis. World J. Gastroenterol. 2009, 15, 2579–2586. [Google Scholar] [CrossRef]
- Wheeler, A.P.; Ridley, A.J. Why three rho proteins? Rhoa, rhob, rhoc, and cell motility. Exp. Cell Res. 2004, 301, 43–49. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Log2 Ratio | Protein Accession Numbers | Protein Names | Gene Names | Unique Peptides | Molecular Weight (Da) |
|---|---|---|---|---|---|
| 2.44 | Q6FHZ7 | Perilipin-2 | ADFP | 14 | 48,075 |
| 1.83 | Q53GP1 | N-sulfoglucosamine sulfohydrolase | SGSH | 2 | 56,642 |
| 1.09 | P81605 | Dermcidin | DCD | 4 | 11,284 |
| 0.89 | J3KPX7 | Prohibitin-2 | PHB2 | 8 | 33,239 |
| 0.83 | B2R4R9 | 40S ribosomal protein S28 | RPS28 | 2 | 78,409 |
| 0.81 | Q9H477 | Ribokinase | RBKS | 2 | 34,143 |
| 0.76 | A0A024RA32 | Glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferase 1 | C1GALT1 | 2 | 42,202 |
| 0.76 | Q53H37 | Calmodulin-like protein 5 | CALML5 | 4 | 15,876 |
| 0.73 | Q5QNW6 | Histone H2B type 2-F | HIST2H2BF | 5 | 13,920 |
| 0.70 | A0A024R6I3 | Transmembrane emp24 domain-containing protein 10 | TMED10 | 3 | 24,976 |
| 0.70 | Q71UI9 | Histone H2A.V | H2AFV | 4 | 13,509 |
| 0.70 | Q7Z7M4 | Superoxide dismutase; mitochondrial | SOD2 | 5 | 23,672 |
| −0.81 | F6XU50 | Lymphoid-specific helicase | HELLS | 6 | 82,732 |
| −0.82 | B4DTD8 | Glypican-3; secreted glypican-3 | GPC3 | 3 | 63,727 |
| −0.83 | A7BI36 | Ribosome-binding protein 1 | RRBP1 | 83 | 165,750 |
| −0.83 | A0A1L5BXV2 | Receptor expression-enhancing protein | REEP6 | 2 | 20,733 |
| −0.87 | Q53G72 | B-cell receptor-associated protein 31 | BCAP31 | 4 | 27,931 |
| −1.11 | B3KPF6 | Striatin-4 | STRN4 | 3 | 38,960 |
| −1.20 | Q96IF1 | LIM domain-containing protein ajuba | AJUBA | 5 | 56,933 |
| −1.46 | B4DZM3 | Ribosomal RNA processing protein 1 | RRP1 | 3 | 46,150 |
| Ingenuity Canonical Pathways | p-Value | z-Score | Molecules |
|---|---|---|---|
| Unfolded protein response | 1.32 × 10−6 | −2.44 | CALR,HSP90B1,HSPA5,P4HB,PDIA6,UBXN4 |
| Protein kinase A signaling | 9.12 × 10−4 | −2.33 | CALML5,H1-2,H1-4,H1 5,MYH10,MYL12A,PDIA3,PRKAR1A,RHOA |
| Xenobiotic metabolism CAR signaling pathway | 3.09 × 10−2 | −2 | ALDH3A2,GSTZ1,HSP90B1,MGST1 |
| HER-2 signaling in breast cancer | 3.31 × 10−2 | −2 | ARF4,COX6B1,RPS6,YES1 |
| Insulin secretion signaling pathway | 6.61 × 10−2 | −2 | PDIA3,PRKAR1A,SSR4,YES1 |
| Xenobiotic metabolism PXR signaling pathway | 2.04 × 10−4 | −1.89 | ALDH3A2,CES1,CES2,GSTZ1,HSP90B1,MGST1,PRKAR1A |
| Sirtuin signaling pathway | 3.55 × 10−2 | −1.34 | H1-2,H1-4,H1-5,PRKDC,SOD2 |
| Cardiac hypertrophy signaling (enhanced) | 1.98 × 10−1 | −1.34 | CALML5,PDIA3,PRKAR1A,RHOA,RPS6 |
| Xenobiotic metabolism AHR signaling pathway | 1.95 × 10−3 | −1 | ALDH3A2,GSTZ1,HSP90B1,MGST1 |
| Opioid signaling pathway | 6.92 × 10−2 | −1 | AP1B1,CALML5,PRKAR1A,YES1 |
| Integrin signaling | 1.07 × 10−2 | −0.44 | ACTG1,ARF4,CAPNS1,MYL12A,RHOA |
| Cardiac hypertrophy signaling | 1.74 × 10−2 | −0.44 | CALML5,MYL12A,PDIA3,PRKAR1A,RHOA |
| Synaptogenesis signaling pathway | 4.57 × 10−2 | −0.44 | AP1B1,CALML5,PRKAR1A,RHOA,YES1 |
| Estrogen receptor signaling | 1.07 × 10−3 | −0.37 | HNRNPD,HSP90B1,MYL12A,PDIA3,PRKAR1A,PRKDC,RHOA,SOD2 |
| ILK signaling | 6.76 × 10−3 | 0.44 | ACTG1,KRT18,MYH10,MYH9,RHOA |
| Actin cytoskeleton signaling | 1.38 × 10−2 | 0.44 | ACTG1,MYH10,MYH9,MYL12A,RHOA |
| Hepatic fibrosis signaling pathway | 9.55 × 10−3 | 1.13 | CALML5,MYL12A,PRKAR1A,RHOA,SOD2,TFRC,YAP1 |
| Ferroptosis signaling pathway | 1.38 × 10−4 | 1.63 | ARF4,FDFT1,H2AX,SLC39A14,TFRC,YAP1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altomare, A.A.; Aiello, G.; Garcia, J.L.; Garrone, G.; Zoanni, B.; Carini, M.; Aldini, G.; D’Amato, A. Protein Profiling of a Cellular Model of NAFLD by Advanced Bioanalytical Approaches. Int. J. Mol. Sci. 2022, 23, 9025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169025
Altomare AA, Aiello G, Garcia JL, Garrone G, Zoanni B, Carini M, Aldini G, D’Amato A. Protein Profiling of a Cellular Model of NAFLD by Advanced Bioanalytical Approaches. International Journal of Molecular Sciences. 2022; 23(16):9025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169025
Chicago/Turabian StyleAltomare, Alessandra Anna, Gilda Aiello, Jessica Leite Garcia, Giulia Garrone, Beatrice Zoanni, Marina Carini, Giancarlo Aldini, and Alfonsina D’Amato. 2022. "Protein Profiling of a Cellular Model of NAFLD by Advanced Bioanalytical Approaches" International Journal of Molecular Sciences 23, no. 16: 9025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169025