1. Introduction
Pathogenic variants in
EFTUD2, which encodes a GTPase and a core component of the U5 subunit of the spliceosome [
1], are responsible for the congenital syndrome Mandibulofacial Dysostosis with Microcephaly (MFDM) (OMIM#610536). MFDM patients have a range of craniofacial abnormalities that include micrognathia, small dysplastic pinnae, malar hypoplasia, hearing loss, microcephaly and developmental delay [
2]. Mutant mice carrying a heterozygous loss-of-function mutation in
Eftud2 are viable and fertile, and do not model MFDM [
3]. On the other hand, embryos that are homozygous for the same loss of function mutation fail to implant and arrest at the blastocyst stage [
3]. However,
Eftud2ncc−/− embryos with
Wnt1-Cre2 mediated deletion of exon 2 of
Eftud2 have severe brain and craniofacial malformations, in the same precursor that is affected in MFDM patients, thereby modelling those head and face defects that are found in patients [
4].
RNAseq analysis using heads of
Eftud2ncc−/− embryos, prior to the onset of malformations, revealed increased skipping of exon 3 of
Mdm2, a master regulator of
Trp53 (P53). Additionally, the levels of nuclear P53 and P53-regulated genes, including
Mdm2, were increased in
Eftud2ncc−/− embryos and in O9-1 neural crest cells after siRNA mediated knockdown of
Eftud2. Furthermore, levels of P53 target genes were reduced when full-length
Mdm2 was overexpressed in O9-1 cells with
Eftud2 knockdown, and treatment of pregnant females with pifithrin-α improved craniofacial development in
Eftud2ncc−/− embryos [
4]. Therefore, we proposed that craniofacial malformations that are found in
Eftud2ncc−/− embryos was a consequence of the mis-splicing of
Mdm2 leading to hyperactivation of the P53 pathway.
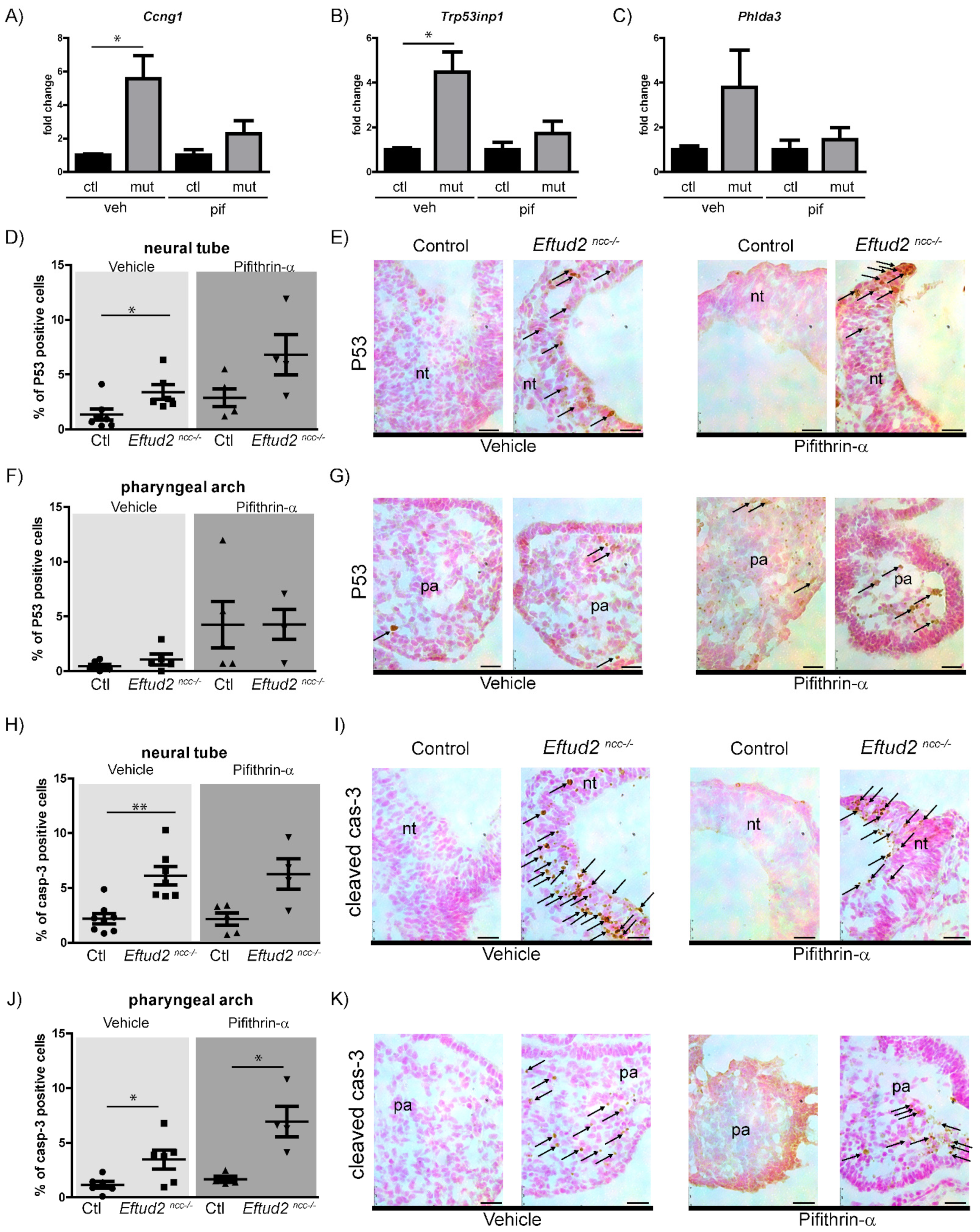
Herein, we show that P53 level and activity are reduced in Eftud2ncc−/− embryos that were treated with pifithrin-α. These embryos also had reduced apoptosis in their neural tube. We generated Eftud2ncc−/−; Trp53ncc−/− double mutant embryos and showed that they had reduced nuclear P53 and P53-activity, no changes in mitosis, and reduced apoptosis in their neural tube. Furthermore, although Eftud2ncc−/−; Trp53ncc−/− mutants have more SOX10 positive cranial neural crest cells, when compared to Eftud2ncc−/− (Eftud2ncc−/−; Trp53 +/+) embryos, they are morphologically abnormal from embryonic day (E)9.5 to E14.5 and they die before birth, similar to the Eftud2ncc−/− mutants. Additionally, Eftud2ncc−/−; Trp53ncc−/− embryos have a significant increase in exon-skipping in P53-regulated transcripts, including Mdm2 and FoxM1, and a non-significant increase in levels of the P53-independent transcript Synj2bp, when compared to controls or Eftud2ncc−/− mutants. Our data indicate that P53 hyperactivation contributes to apoptosis in the neural tube and suggests that P53 attenuates mis-splicing in Eftud2 mutant cells. Notably our study shows that craniofacial malformations and death of Eftud2ncc−/− mutant embryos are independent of P53-activation.
3. Discussion
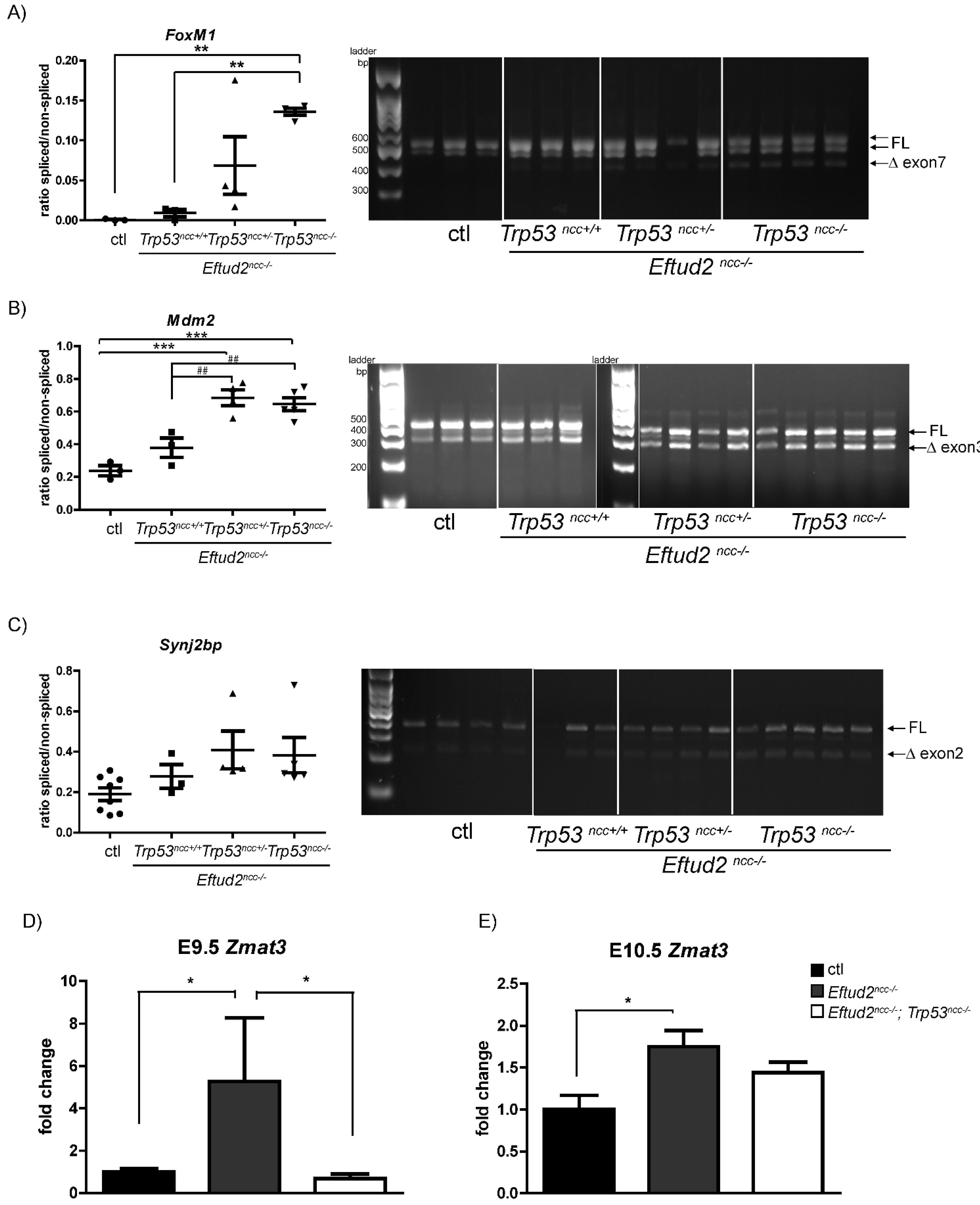
Herein, we investigated the contribution of the P53-pathway to craniofacial malformations in Eftud2ncc−/− mutant embryos. We show that we can reduce P53 levels and activity in the neural tube by treating Eftud2ncc−/− embryos with pifithrin-α or using Wnt1-Cre2 to delete both alleles of Trp53. Furthermore, although reducing P53-activity attenuated apoptosis in the neural tube of Eftud2ncc−/− embryos, it did not rescue craniofacial development. We also show that mis-splicing is increased in Eftud2ncc−/−; Trp53ncc−/− mutants and that this increase was not associated with a significant change in levels of the P53-regulated splicing factor, Zmat3. These data indicate that P53-independent pathways contribute to craniofacial defects in Eftud2ncc−/− mutants and reveal a previously unappreciated role for P53 in regulating mis-splicing.
The increased accumulation and activity of P53 secondary to mutations in genes that are involved in a diverse array of cellular processes, including splicing, contribute to malformations in animal models of developmental syndromes [
10,
11]. In a few cases, increased P53 activity was also found in clinical samples from patients with developmental syndromes [
11]. More specifically, mouse models of neurocristopathies have overactivation of the P53 pathway and reducing the levels of P53 improve and in some cases rescue malformations in these models [
12,
13]. A similar partial rescue was found when
Eftud2 was mutated in zebrafish [
14,
15]. Based on our previous findings that pifithrin-α improved neural tube and craniofacial development in
Eftud2ncc−/− embryos we postulated that reducing P53 in mouse would lead to a similar outcome [
4].
In the present study, we show that P53-activity was increased in the heads of E9.5
Eftud2ncc−/− embryos and that this increase was attenuated by pifithrin-α. More specifically, we show that
Eftud2ncc−/− mutant embryos at this stage had increased nuclear P53 accumulation and apoptosis in the neural tube, but not the first pharyngeal arch. Therefore, and not surprisingly, treating these mutant embryos with pifithrin-α led to reduced apoptosis in the neural tube and not in the first pharyngeal arch. We previously showed that the perimeter of the first pharyngeal arch of pifithrin-α treated
Eftud2ncc−/− embryos is larger than those of vehicle-treated mutants [
4]. Therefore, it is surprising that we found a significant increase in apoptosis in pharyngeal arches of embryos that were exposed to pifithrin-α from E6.5 to E8.5. Although this data suggest that P53-independent activity leads to the death of
Eftud2 mutant cells in the pharyngeal arches, further work is needed to decipher how a loss of
Eftud2 results in abnormal development of the first arch and its derivatives, including Meckel’s cartilage. Although first identified as a P53 inhibitor, pifithrin-α can also have P53-independent activity [
16]. In fact, its mechanism of action remains unclear and its specific effect on P53 may vary in different cell types. For instance, pifithrin-α was associated with cell-type specific phosphorylation of P53 and the differential expression of its target genes [
16]. Moreover, it was reported to reduce intracellular reactive oxygen species (ROS) independently of P53 [
16]. To rule out the contribution of a P53-independent mechanisms to the partial rescue of craniofacial development that were found in pifithrin-α-treated
Eftud2ncc−/− embryos, we used a genetic approach and deleted
Trp53 in
Eftud2 mutant cells.
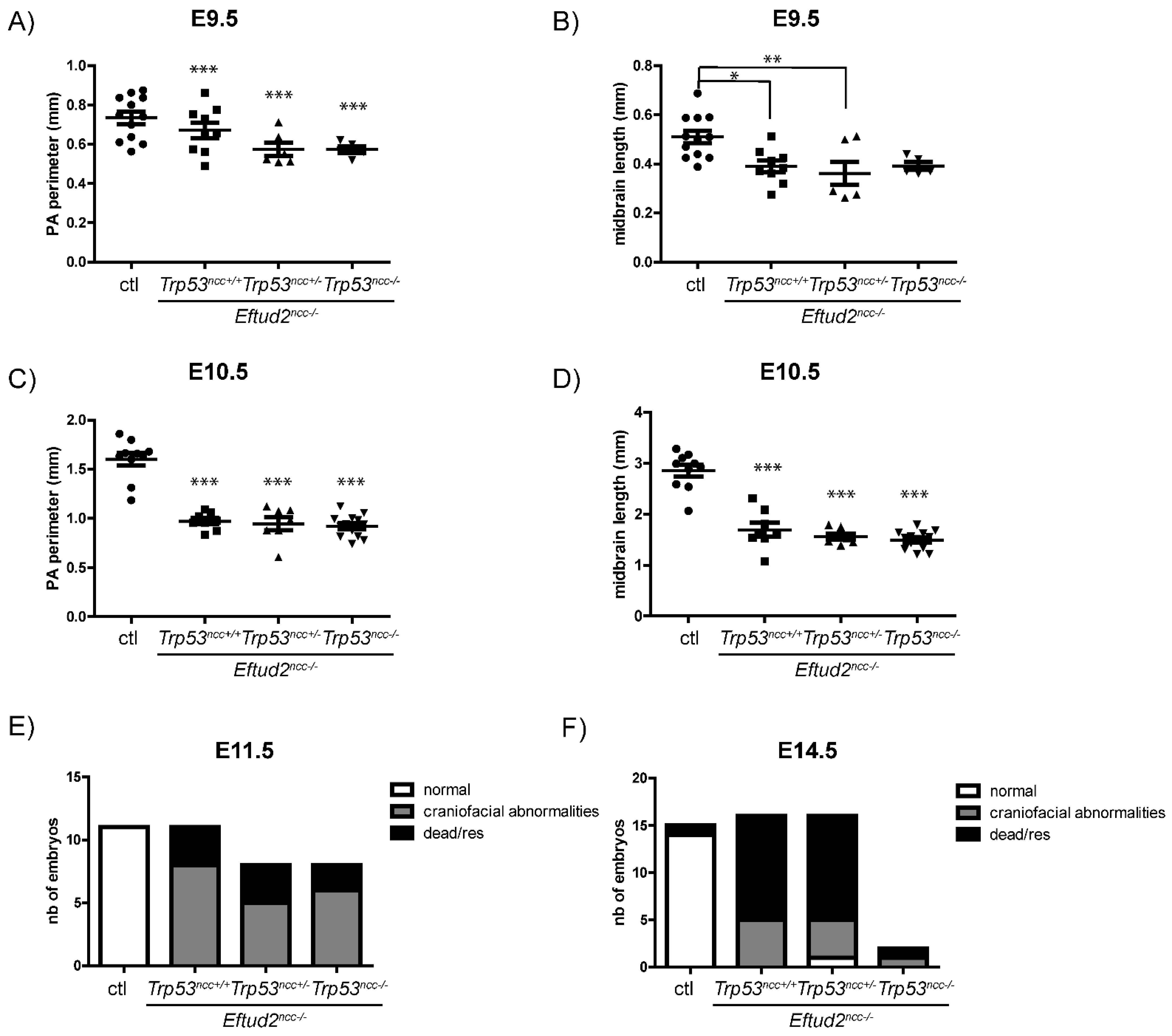
When we used the Wnt1-Cre2 transgenic mouse line to remove both alleles of Eftud2 and one allele of Trp53 (Eftud2ncc−/−; Trp53ncc+/− embryos), there was still an increase in nuclear P53 accumulation and in P53 activity. Additionally, most Eftud2ncc−/−; Trp53nc+/− embryos resembled Eftud2ncc−/− mutants, except for one morphologically normal embryo that was recovered at E14.5. Therefore, we cannot completely rule out the possibility that reducing the levels of P53 can prevent craniofacial defects in Eftud2ncc−/− mutants. However, this is likely to be a very rare event. In fact, when all stages were combined, no additional morphologically normal Eftud2ncc−/−; Trp53ncc+/− (n = 42) or Eftud2ncc−/−; Trp53ncc−/− (n = 35) embryos were found. Unfortunately, molecular studies could not be performed on this embryo as the litter was collected for cartilage preparations.
Since Eftud2 and Trp53 both map to chromosome 11 and are only 33cM apart, it was difficult to generate both Eftud2ncc−/− and Eftud2ncc−/−; Trp53ncc−/− embryos from the same cross. Therefore, we generated Eftud2ncc−/−; Trp53ncc+/− and Eftud2ncc−/−; Trp53ncc−/− embryos in separate crosses and compared the morphological and the molecular defects that were found to those that we previously described in Eftud2ncc−/− embryos. Our current studies show that P53 accumulation and activity were reduced in Eftud2ncc−/−; Trp53ncc−/− mutant embryos, suggesting that the P53-pathway was efficiently inhibited. Furthermore, as was found for pifithrin-α treated Eftud2ncc−/− mutants, apoptosis was significantly reduced in the neural tube of double homozygous mutant embryos. However, in double homozygous mutant embryos the onset of craniofacial malformations, and the size of the pharyngeal arches and the midbrain, were no different from Eftud2ncc−/− embryos wild type for Trp53. Nor did a loss of P53 extend the life of Eftud2ncc−/− embryos. Altogether, these data indicate that the malformations found in Eftud2ncc−/− embryos are predominantly P53-independent. In the future, we will identify the pathways that are responsible for these malformations.
Nonetheless, our study suggests that the reduced apoptosis in the neural tube most likely leads to an increase in the number of neural crest cells in
Eftud2ncc−/−;
Trp53ncc−/− embryos. Supporting this, we found reduced SOX10 expression in E9.0
Eftud2ncc−/− mutants that were morphologically similar to controls. Moreover, removing both copies of
Trp53 resulted in an increase of SOX10 expression in the head of
Eftud2ncc−/−;
Trp53ncc−/− embryos. Using the ROSA26R reporter, we previously showed that the proportion of Cre-expressing cells was similar in control and E9.0
Eftud2ncc−/− mutant embryos, suggesting that these embryos have a similar number of neural crest cells [
4]. Our results from this study indicate that at this stage,
Eftud2ncc−/− mutant cranial neural crest cells have attenuated expression of SOX10, a marker of post-migratory neural crest cells. Thus, we propose that increased P53 activity in the cranial neural crest cells of E9.0
Eftud2ncc−/− mutants as they exit the neural tube leads to the abnormal expression of proteins such as SOX10, important for their survival and patterning in the pharyngeal arches. Since removing P53 increases expression of SOX10 in
Eftud2ncc−/− mutant cells but does not rescue craniofacial development, we further postulate that SOX10 is not sufficient to protect
Eftud2ncc−/− mutant neural crest cells in the first pharyngeal arch from undergoing cell death at E9.5. However, although we assumed that the number of surviving neural crest cells is insufficient to rescue brain and craniofacial development, we cannot rule out the possibility that surviving neural crest cells cannot differentiate and form the cartilage and bones of the head and face. Furthermore, although we did not see increased splicing of
Xbp1 suggesting that the IRE1α pathway was not activated, we cannot exclude that one of the remaining two- arms of the ER-stress pathway is activated and contributes to Caspase-3 independent cell death of
Eftud2ncc−/− mutant cells.
We next examined our RNAseq dataset for additional pathways/transcripts which may contribute to defects. We examined a role for
FoxM1, a transcription factor which is essential for G2 progression into mitosis [
17], that was mis-spliced in these mutants.
FoxM1 is required for embryonic survival and normal craniofacial development [
18], and is expressed in the head and the pharyngeal arches of E10.5 embryos [
19]. However, its expression and that of its downstream target,
Cdc25b, was not significantly changed in mutant embryos, ruling it out as a major contributor to craniofacial defects found in
Eftud2ncc−/− embryos that were wild-type or mutant for
Trp53.Surprisingly, the mis-splicing of at least three-transcripts:
Foxm1,
Mdm2 and
Snynj2b was increased in
Eftud2ncc−/−;
Trp53ncc−/− embryos, suggesting that wild-type P53 activity tampered mis-splicing. Therefore, we examined the expression of
Zmat3 which encodes for an RNA binding protein that is regulated by P53 [
9]. We hypothesized that the expression of this splicing factor would be further increased in
Eftud2ncc−/−;
Trp53ncc−/− embryos, thus explaining the increased exon skipping that was found. However,
Zmat3 overexpression was completely abrogated when
Trp53 was deleted, consistent with the observation that it is regulated by P53 [
9]. We propose that one, or more, yet to be identified splicing factors are negatively regulated by P53 and are over expressed in
Eftud2ncc−/−;
Trp53ncc−/− embryos.
Identifying this gene(s) will be essential for determining how P53 attenuates splicing of Foxm1, Mdm2 and Snynj2b in Eftud2 mutant cells. Further RNAseq analysis using the heads of double mutant embryos will be needed to confirm a global increase in mis-splicing.
4. Materials and Methods
4.1. Mouse Lines
All the procedures and experiments were performed according to the guidelines of the Canadian Council on Animal Care and approved by the Animal Care Committee of the Montreal Children’s Hospital. Wild-type CD1 mice (strain code 022) were purchased from Charles Rivers (Laval, QC, Canada) and wild-type C57Bl/6 mice (stock #000664) were purchased from Jackson Laboratories (Augusta, ME, USA).
Wnt1-Cre2 mice on the 129S4 genetic background were purchased from Jackson’s laboratory (stock# 022137). These
Wnt1-Cre2 transgenic mice express Cre recombinase under the control of the mouse
Wnt1, wingless-related MMTV integration site 1, promoter and enhancer [
20]. The
Trp53tm1brn mouse line on the C57BL/6 genetic background with
loxP sites flanking exons 2–10 of the
Trp53 gene was purchased from Jackson’s laboratory (
Trp53loxP/+) (stock# 008462) [
5]. The generation and the characterization of the conditional
Eftud2em2Lajm (
Eftud2loxp/+) line on the inbred C57BL/6 and mixed CD1 genetic backgrounds, and the
Eftud2em1Lajm (
Eftud2+/−) exon 2 deletion line on a mixed CD1 genetic background were described previously [
3,
4].
4.2. Pifithrin-α Treatment
Pregnant females from mating between
Eftud2loxP/loxP and
Eftud2+/−;
Wnt1- Cre2tg/+ mice were injected with 2.2mg/kg pifithrin-α (Sigma-Aldrich, Saint-Louis, Missouri, USA) or 2% DMSO/PBS (vehicle) daily through intra-peritoneal injection, starting at E6.5 until E8.5. The embryos that were collected at E9.5 were previously generated [
4,
12].
4.3. Generation of Mutation in Eftud2 and Trp53 in Neural Crest Cell-Specific
Trp53 and
Eftud2 both map to mouse chromosome 11 and are approximately 33cM apart. Since these two genes show non-mendelian segregation (
Table S1),
Eftud2ncc−/−;
Trp53ncc+/− (
Eftud2loxP/−;
Trp53loxP/+;
Wnt1-Cre2tg/+) embryos were generated by mating
Eftud2loxP/+;
Trp53loxp/loxp and
Eftud2+/−;
Wnt1-Cre2tg/+ mice. We generated double homozygous mutant embryos (
Eftud2loxP/−;
Trp53loxp/loxp;
Wnt1-Cre2tg/+) by mating
Eftud2loxP/loxP;
Trp53loxp/loxp mice to
Eftud2+/−;
Trp53loxP/loxP;
Wnt1-Cre2tg/+ mice.
Eftud2ncc−/− (
Eftud2loxP/−;
Wnt1-Cre2 tg/+) mutant embryos that were generated as previously described were used for comparison [
4]. From these matings, we used the embryos that did not carry the
Wnt1-Cre2 transgene as controls.
Cre recombinase activity was reported in the male germline of
Wnt1-Cre2 mice on the 129S4 background [
21]. Since the embryos carrying homozygous mutation of
Eftud2 in all cells arrest pre-implantation, [
3] we did not recover
Eftud2ncc−/− embryos with germline homozygous deletion of
Eftud2. All embryos that were analyzed in this study were on a mixed genetic background following the multiple crosses that were needed to generate double homozygous mutants.
4.4. Collection of Embryos
For embryo collection, the day that a vaginal plug was seen was considered embryonic day 0.5 (E0.5). On the day of dissection, the embryos were removed from their extraembryonic membranes; for stages E8.5 to E10.5, the number of somites was counted under light microscope (Leica MZ6 Infinity1 stereomicroscope). The embryos were fixed in 1 or 4% paraformaldehyde at 4 °C overnight (unless otherwise stated), washed in PBS and kept at 4 °C. The yolk sacs were collected and used for genomic DNA extraction for genotyping.
4.5. Cartilage Preparation
To evaluate cartilage formation, embryos were stained with Alcian Blue as previously described [
4]. BABB-cleared embryos were visualized under a light microscope (Leica MZ6 Infinity1 stereomicroscope).
4.6. Preparation of Embryos for Embedding and Histology
The dissected embryos were fixed in 1% paraformaldehyde overnight. For cryo-embedding, the fixed embryos were first cryoprotected in 30% sucrose overnight, embedded in cryomatrix and sectioned at 10 μm thickness for immunohistochemistry and immunofluorescence.
4.7. Immunohistochemistry (IHC) and Immunofluorescence (IF)
Cleaved Caspase-3 (1:250, cat#9661T, Cell Signaling, NEB, Whitby, Ontario, Canada), P53 (1:250, cat#2524, Cell Signaling, NEB, Whitby, ON, Canada), and Phosphohistone H3 (Ser10) (1:200, cat#06-570, Sigma-Aldrich, Saint-Louis, MO, USA) primary antibodies were used. For IHC, an avidin/biotin-peroxidase based system was used (VECTASTAIN® Elite ABC HRP Kit: PK-620000, Vector Laboratories, Newark, CA, USA) containing biotinylated universal (anti-mouse/rabbit IgG) secondary antibody and visualized with DAB (Vector Laboratories). After rinsing with water, the slides were counterstained with Nuclear Fast Red before mounting with an aqueous mounting medium. For quantification of colorimetric signal, particle analysis on Image J was used. 2 to 3 sections per embryo were imaged. To count the number of total cells, the sections after the ones that were used for IHC was mounted with DAPI and particle analysis on Image J was used to determine the total number of cells in the head. The percentage of positive cells were determined as follows: the number of P53 or cleaved Caspase-3-positive cells/number of DAPI-positive cells in the head X100, and plotted using GraphPad (Prism).
For wholemount immunofluorescence, embryos were fixed in 1%PFA overnight, washed with PBS and incubated with SOX10 antibody (1:100, cat#78330S, Cell Signaling, NEB, Whitby, ON, Canada) in blocking buffer (1% BSA, 5% serum, 0.3% TritonX100) overnight at 4 °C. After washing with PBS, embryos were incubated with goat anti-rabbit Alexa 568 (1:500, #A11011, ThermoFisher, Waltham, MA, USA) in blocking buffer overnight with DAPI (1 μg/mL). On the next day, the embryos were washed, and images were captured on Leica microsystem (model DM6000B) and Leica camera (model DFC 450). We thank Dr. Colin Dinsmore for the protocol.
4.8. RNA Isolation for RT-qPCR
RNA extraction and RT-qPCR analysis was performed as previously published [
4]. The primers used are listed in
Table 1.
4.9. Primers Used for Splicing Analysis
For
Mdm2,
FoxM1 and
Synj2bp splicing analysis, cDNA was amplified with a RT-PCR program that included a hot start at 95 °C for 5 min, followed by 35 cycles of a denaturation step at 95 °C for 10 s, an annealing step at 55 °C for 30 s, an extension step at 72 °C for 45 s with a final extension step at 72 °C for 10 min. The products were visualized on a 2% agarose gel. The primers that were used are as follow:
Mdm2 Forward (exon2-6/7): GATCACCGCGCTTCTCCTGC,
Mdm2 Reverse (exon2-6/7): GATGTGCCAGAGTCTTGCTG [
22];
FoxM1 forward: CTTTAAGCACATTGCCAAGC.
FoxM1 reverse: GGTTCTGTGGCAGGAAGC; Synjbp2 forward: CGGAGGAAGAGATCAACCTG, Synjbp2 reverse: TATCTCACGAAGGCCCAAAC.
The intensity of the bands was quantified using Image J and the ratios of spliced vs. non-spliced were plotted by GraphPad Prism.
4.10. ER Stress
To determine if the unfolded protein response pathway is activated in
Eftud2ncc−/−;
Trp53ncc+/− or
Eftud2ncc−/−;
Trp53ncc−/− embryos, we used primers that were previously designed to amplify a 350 bp portion of
Xbp1 (Forward primer: GATCCTGACGAGGTTCCAGA and Reverse primer: GGTCCCCACTGACAGAGAAA), followed by digestion of the amplicon with
Pst1 restriction enzyme, as previously described [
23]. Since
Pst1 does not cut the amplicon in the absence of ER stress, the presence of ER stress is indicated by the presence of a 240 bp and a 110 bp bands.
4.11. Statistical Analysis
Two-tailed non-parametric Mann-Whitney t-test analysis was performed using Excel and Prism Software. ANOVA test analysis followed by Tukey’s post-test to compare all pairs of columns and a Chi-square test were performed using Prism. Significant p-values are represented as * p < 0.05, ** p < 0.01 and *** p < 0.001. All unique/stable reagents that were generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}