
Hypothermia Alleviates Reductive Stress, a Root Cause of Ischemia Reperfusion Injury
 and
and 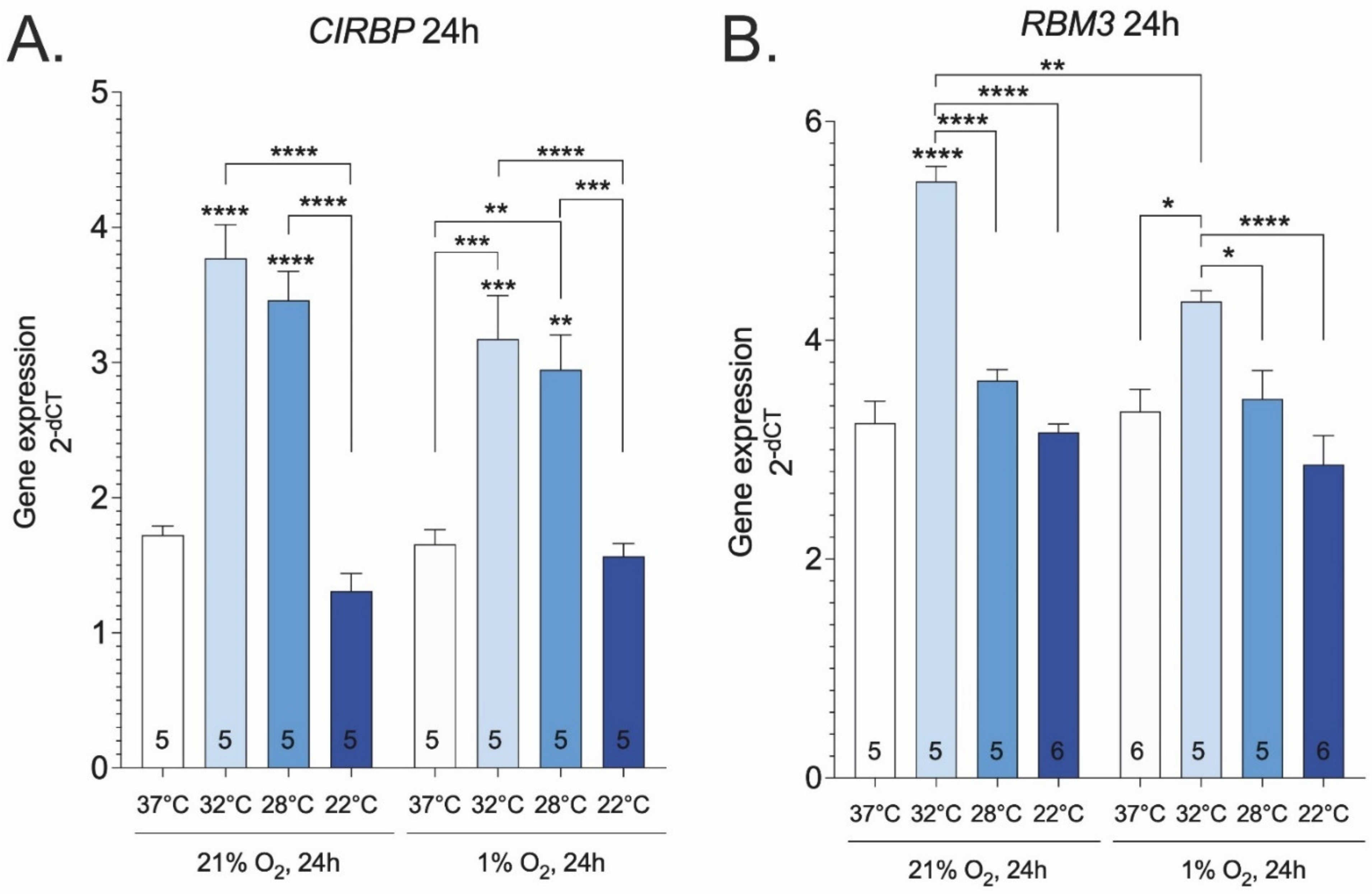{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
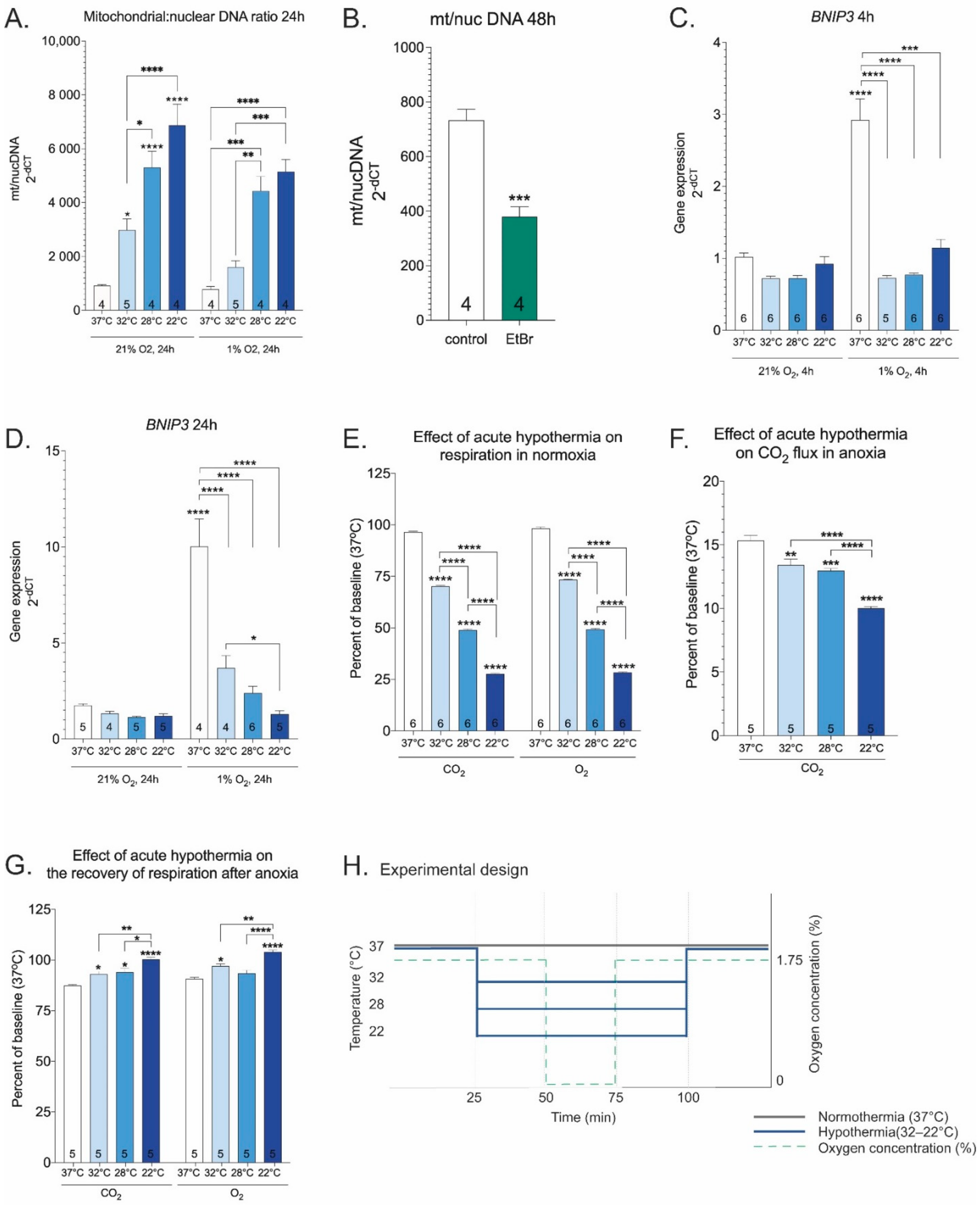
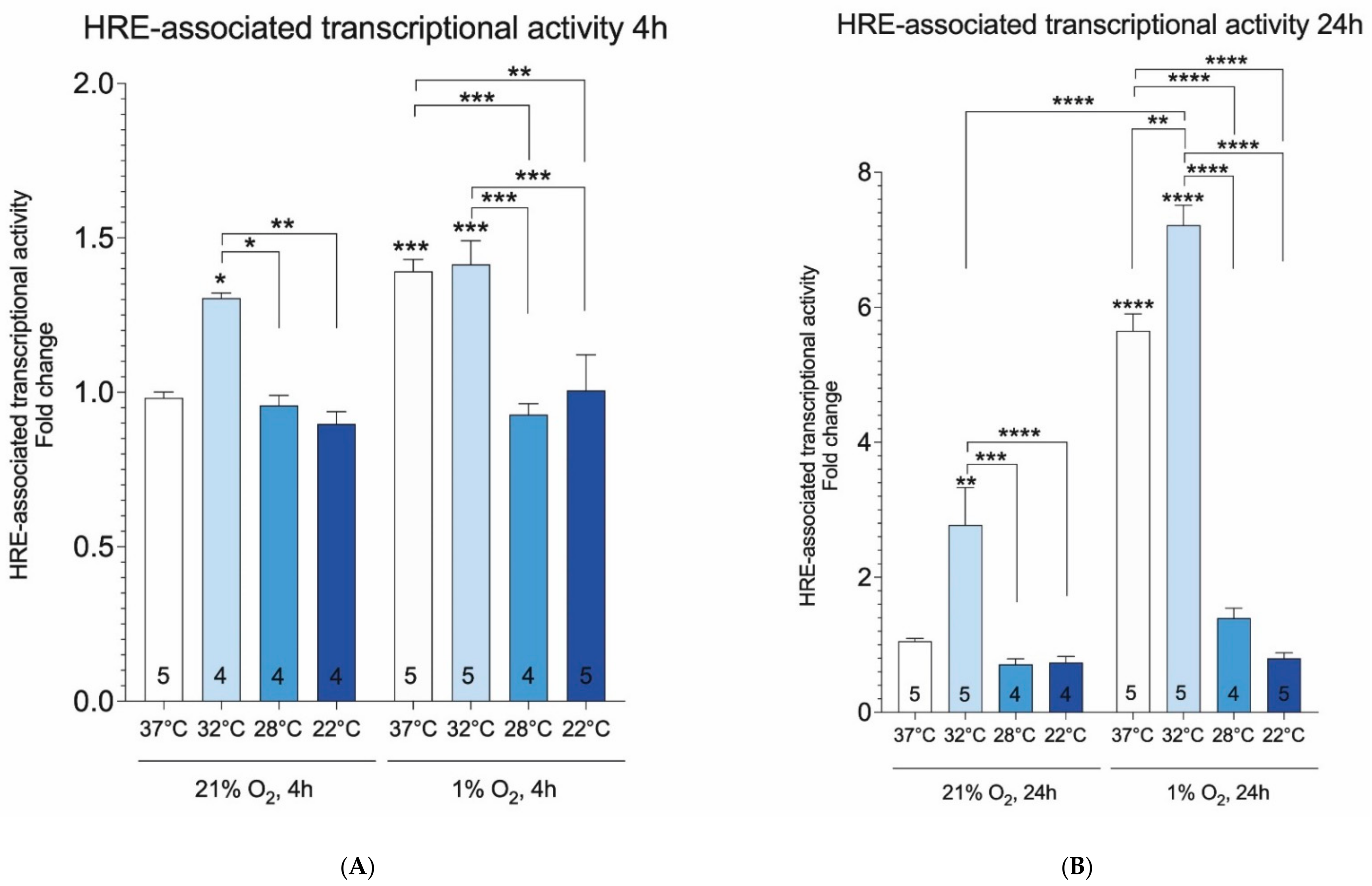
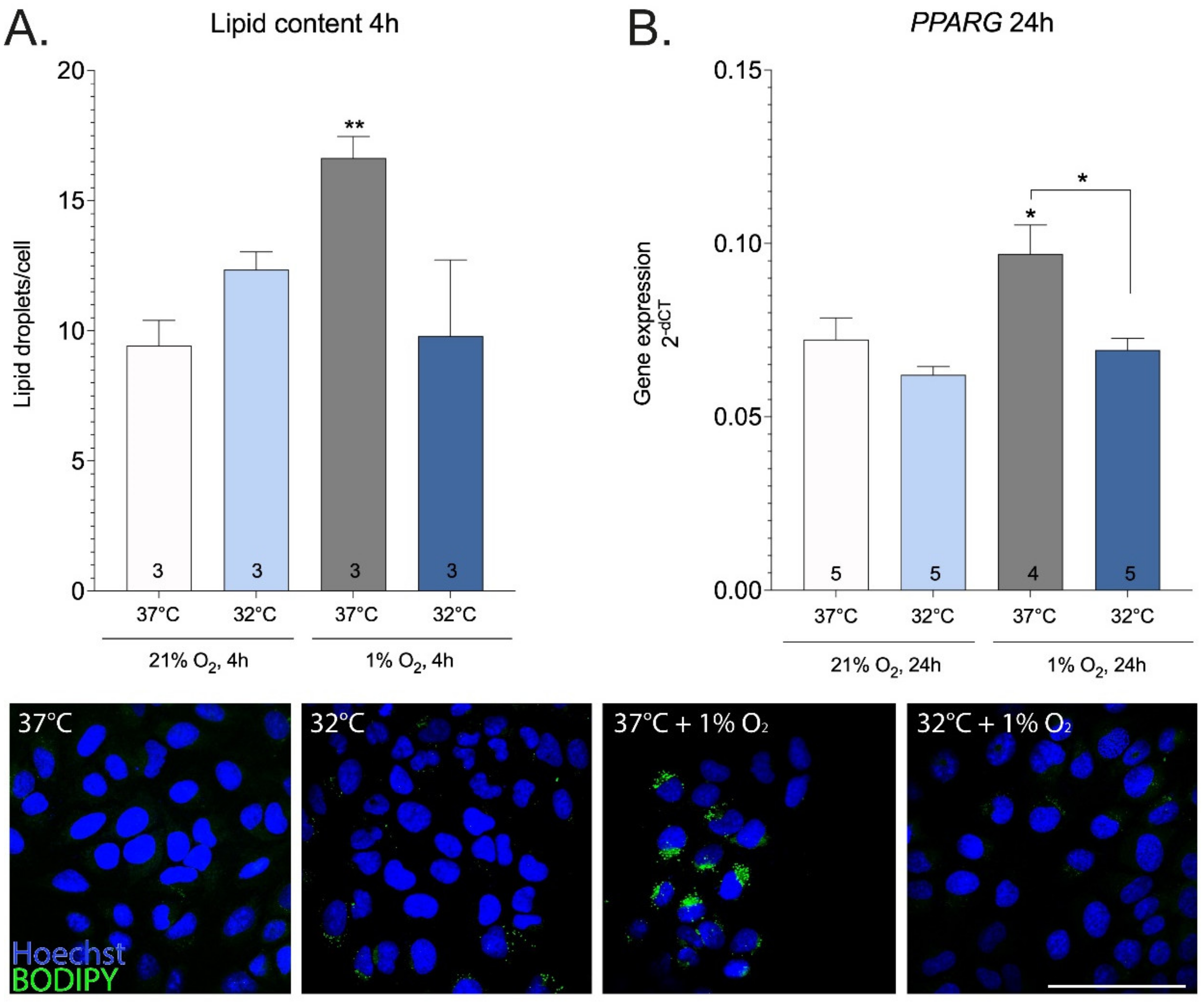
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Treatments
4.3. Luciferase-Reporter Assay
4.4. RNA Extraction
4.5. DNA Extraction
4.6. Quantitative Real-Time Reverse Transcription PCR
4.7. ADP/ATP Ratio Assay Kit
4.8. Metabolomics
4.9. Lipid Content
4.10. Microscopy and Image Analysis
4.11. Cell Respiration Experiments
4.12. Optical Absorption Measurements
4.13. Gas System for Simultaneous Measurements of Respiratory CO2 and O2 Exchange
4.14. O2 Concentration Calculations
4.15. Respirometric Analysis with Oroboros
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krezdorn, N.; Tasigiorgos, S.; Wo, L.; Turk, M.; Lopdrup, R.; Kiwanuka, H.; Win, T.-S.; Bueno, E.; Pomahac, B. Tissue conservation for transplantation. Innov. Surg. Sci. 2017, 2, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Moers, C.; Pirenne, J.; Paul, A.; Ploeg, R.J. Machine Perfusion or Cold Storage in Deceased-Donor Kidney Transplantation. N. Engl. J. Med. 2012, 366, 770–771. [Google Scholar] [CrossRef]
- Taylor, M.J.; Baicu, S.C. Current state of hypothermic machine perfusion preservation of organs: The clinical perspective. Cryobiology 2010, 60, S20–S35. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, L.; Daloze, P.M.; Huguet, C.; Groth, C.G.; Kashiwagi, N.; Hutchison, D.E.; Starzl, T.E. Successful orthotopic transplantation of liver homografts after eight to twenty-five hours preservation. Surg. Forum 1967, 18, 376–378. [Google Scholar] [PubMed]
- Van Rijn, R.; Schurink, I.J.; de Vries, Y.; Berg, A.P.V.D.; Cerisuelo, M.C.; Murad, S.D.; Erdmann, J.I.; Gilbo, N.; de Haas, R.J.; Heaton, N.; et al. Hypothermic Machine Perfusion in Liver Transplantation-A Randomized Trial. N. Engl. J. Med. 2021, 384, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Yao, L.; Zhao, M.; Peng, L.-P.; Liu, M. Organ preservation: From the past to the future. Acta Pharmacol. Sin. 2018, 39, 845–857. [Google Scholar] [CrossRef]
- Zanetto, A.; Russo, F.P.; Germani, G.; Burra, P. Organ Preservation in Liver Transplantation. Semin. Liver Dis. 2018, 38, 260–269. [Google Scholar] [CrossRef]
- Vairetti, M.; Ferrigno, A.; Carlucci, F.; Tabucchi, A.; Rizzo, V.; Boncompagni, E.; Neri, D.; Gringeri, E.; Freitas, I.; Cillo, U. Subnormothermic machine perfusion protects steatotic livers against preservation injury: A potential for donor pool increase? Liver Transplant. 2009, 15, 20–29. [Google Scholar] [CrossRef]
- Carrel, A.; Lindbergh, C.A. The Culture of Whole Organs. Science 1935, 81, 621–623. [Google Scholar] [CrossRef]
- Collins, G.; Bravo-Shugarman, M.; Terasaki, P. Kidney preservation for transportation: Initial Perfusion and 30 Hours’ Ice Storage. Lancet 1969, 294, 1219–1222. [Google Scholar] [CrossRef]
- Jahania, M.; Sanchez, J.; Narayan, P.; Lasley, R.D.; Mentzer, R.M. Heart preservation for transplantation: Principles and strategies. Ann. Thorac. Surg. 1999, 68, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Kosieradzki, M.; Kuczynska, J.; Piwowarska, J.; Wegrowicz-Rebandel, I.; Kwiatkowski, A.; Lisik, W.; Michalak, G.; Danielewicz, R.; Paczek, L.; Rowinski, W.A. Prognostic significance of free radicals: Mediated injury occurring in the kidney donor. Transplantation 2003, 75, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Dhital, K.K.; Iyer, A.; Connellan, M.; Chew, H.C.; Gao, L.; Doyle, A.; Hicks, M.; Kumarasinghe, G.; Soto, C.; Dinale, A.; et al. Adult heart transplantation with distant procurement and ex-vivo preservation of donor hearts after circulatory death: A case series. Lancet 2015, 385, 2585–2591. [Google Scholar] [CrossRef] [PubMed]
- Machuca, T.N.; Mercier, O.; Collaud, S.; Tikkanen, J.; Krueger, T.; Yeung, J.C.; Chen, M.; Azad, S.; Singer, L.; Yasufuku, K.; et al. Lung Transplantation with Donation After Circulatory Determination of Death Donors and the Impact of Ex Vivo Lung Perfusion. Am. J. Transplant. 2015, 15, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Acceptable Ischemic Times. Available online: https://www.nedonation.org/donation-guide/organ/acceptable-ischemic-times (accessed on 24 July 2022).
- Monbaliu, D.; Pirenne, J.; Talbot, D. Liver transplantation using Donation after Cardiac Death donors. J. Hepatol. 2011, 56, 474–485. [Google Scholar] [CrossRef]
- Monbaliu, D.; Brassil, J. Machine perfusion of the liver: Past, present and future. Curr. Opin. Organ Transplant. 2010, 15, 160–166. [Google Scholar] [CrossRef]
- Liu, W.-P.; Humphries, A.L.; Russell, R.; Stoddard, L.D.; Moretz, W.H. 48-Hour Storage of Canine Kidneys after Brief Perfusion with Collinsʼ Solution. Ann. Surg. 1971, 173, 748–757. [Google Scholar] [CrossRef]
- Weissenbacher, A.; Vrakas, G.; Nasralla, D.; Ceresa, C.D.L. The future of organ perfusion and re-conditioning. Transpl. Int. 2019, 32, 586–597. [Google Scholar] [CrossRef]
- Ponticelli, C.E. The impact of cold ischemia time on renal transplant outcome. Kidney Int. 2015, 87, 272–275. [Google Scholar] [CrossRef]
- Talma, N.; Kok, W.; Mestdagh, C.D.V.; Shanbhag, N.; Bouma, H.; Henning, R. Neuroprotective hypothermia-Why keep your head cool during ischemia and reperfusion. Biochim. Biophys. Acta 2016, 1860, 2521–2528. [Google Scholar] [CrossRef]
- Yu, S.P.; Lee, J.H.; Zhang, J. Neuroprotective mechanisms and translational potential of therapeutic hypothermia in the treatment of ischemic stroke. Neural Regen. Res. 2017, 12, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Eskla, K.-L.; Porosk, R.; Reimets, R.; Visnapuu, T.; Vasar, E.; Hundahl, C.A.; Luuk, H. Hypothermia augments stress response in mammalian cells. Free Radic. Biol. Med. 2018, 121, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Tahir, R.; Pabaney, A. Therapeutic hypothermia and ischemic stroke: A literature review. Surg. Neurol. Int. 2016, 7, 381–386. [Google Scholar] [CrossRef]
- Luscombe, M.; Andrzejowski, J.C. Clinical applications of induced hypothermia. Contin. Educ. Anaesth. Crit. Care Pain 2006, 6, 23–27. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Ghyczy, M.; Boros, M. Evidence in support of a concept of reductive stress-Reply by Ghyczy & Boros. Br. J. Nutr. 2002, 87, 94. [Google Scholar]
- Bernard, S.A.; Gray, T.W.; Buist, M.D.; Jones, B.M.; Silvester, W.; Gutteridge, G.; Smith, K. Treatment of Comatose Survivors of Out-of-Hospital Cardiac Arrest with Induced Hypothermia. N. Engl. J. Med. 2002, 346, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N. Engl. J. Med. 2002, 346, 549–556. [Google Scholar]
- Dalen, M.L.; Liu, X.; Elstad, M.; Løberg, E.M. Resuscitation with 100% oxygen increases injury and counteracts the neuroprotective effect of therapeutic hypothermia in the neonatal rat. Pediatr. Res. 2012, 71, 247. [Google Scholar] [CrossRef] [PubMed]
- Bona, E.; Hagberg, H.; Løberg, E.M.; Bågenholm, R. Protective effects of moderate hypothermia after neonatal hypoxia-ischemia: Short- and long-term outcome. Pediatr. Res. 1998, 43, 738. [Google Scholar] [CrossRef]
- Holzer, M. Targeted Temperature Management for Comatose Survivors of Cardiac Arrest. N Engl. J. Med. 2010, 363, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.; Osredkar, D.; Puchades, M.; Maes, E.; Falck, M.; Flatebø, T.; Walløe, L.; Sabir, H.; Thoresen, M. Treatment temperature and insult severity influence the neuro-protective effects of therapeutic hypothermia. Sci. Rep. 2016, 6, 23430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yenari, M.A.; Han, H.S. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat. Rev. Neurosci. 2012, 13, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Jassem, W.; Heaton, N.D. The role of mitochondria in ischemia/reperfusion injury in organ transplantation. Kidney Int. 2004, 66, 514–517. [Google Scholar] [CrossRef]
- Williamson, D.H.; Lund, P.; Krebs, H.A. The redox state of freenicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of ratliver. Biochem. J. 1967, 103, 514–527. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R.-S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Robin, E.D.; Murphy, B.J.; Theodore, J.J. Coordinate regulation of glycolysis by hypoxia in mammalian cells. Cell. Physiol. 1984, 118, 287–290. [Google Scholar] [CrossRef]
- Marti, H.H.; Jung, H.H.; Pfeilschifter, J.; Bauer, C. Hypoxia and cobalt stimulate lactate dehydrogenase (LDH) activity in vascular smooth muscle cells. Eur. J. Physiol. 1994, 429, 216–222. [Google Scholar] [CrossRef]
- Firth, J.D.; Ebert, B.L.; Pugh, C.W.; Ratcliffe, P.J. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: Similarities with the erythropoietin 3′ enhancer. Proc. Natl. Acad. Sci. USA 1994, 91, 6496–6500. [Google Scholar] [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.J.; Brand, M.D. Superoxide production by NADH: Ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004, 382, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant Regulation of Gene Expression by Hypoxia and 2-Oxoglutarate-dependent Dioxygenase Inhibition. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef]
- Forsythe, J.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Tong, G.; Endersfelder, S.; Rosenthal, L.-M.; Wollersheim, S.; Sauer, I.M.; Bührer, C.; Berger, F.; Schmitt, K.R.L. Effects of moderate and deep hypothermia on RNA-binding proteins RBM3 and CIRP expressions in murine hippocampal brain slices. Brain Res. 2013, 1504, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Rzechorzek, N.M.; Connick, P.; Patani, R.; Selvaraj, B.T.; Chandran, S. Hypothermic Preconditioning of Human Cortical Neurons Requires Proteostatic Priming. eBioMedicine 2015, 2, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, H.; Itoh, K.; Kaneko, Y.; Kishishita, M.; Yoshida, O.; Fujita, J. A Glycine-rich RNA-binding Protein Mediating Cold-inducible Suppression of Mammalian Cell Growth. J. Cell Biol. 1997, 137, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Chip, S.; Zelmer, A.; Ogunshola, O.O.; Felderhoff-Mueser, U.; Nitsch, C.; Bührer, C.; Wellmann, S. The RNA-binding protein RBM3 is involved in hypothermia induced neuroprotection. Neurobiol. Dis. 2011, 43, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-T.; Xue, J.-H.; Zhang, Z.-W.; Kong, H.-B.; Liu, A.-J.; Li, S.-C.; Xu, D.-G. Cold-inducible RNA-binding protein inhibits neuron apoptosis through the suppression of mitochondrial apoptosis. Brain Res. 2015, 1622, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Carmichael, J.; Swartz, J.; Muro, S.; Wyttenbach, A.; Matsubara, K.; Rubinsztein, D.C.; Kato, K. Modulation of polyglutamine-induced cell death by genes identified by expression profiling. Hum. Mol. Genet. 2002, 11, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.-H.; Xu, C.-S.; Song, Y.C.; Childs, K.F.; Xiao, Y.; Bolling, S.F.; Lupinetti, F.M.; Portman, M.A. Temperature Threshold and Modulation of Energy Metabolism in the Cardioplegic Arrested Rabbit Heart. Cryobiology 1998, 36, 2–11. [Google Scholar]
- Kanemoto, S.; Matsubara, M.; Noma, M.; Leshnower, B.G.; Parish, L.M.; Jackson, B.M.; Hinmon, R.; Hamamoto, H.; Gorman, J.H.; Gorman, R.C. Mild hypothermia to limit myocardial ischemia-reperfusion injury: Importance of timing. Ann. Thorac. Surg. 2009, 87, 157–163. [Google Scholar]
- Shao, Z.; Sharp, W.W.; Wojcik, K.R.; Li, C.; Han, M.; Chang, W.; Ramachandran, S.; Li, J.; Hamann, K.J.; Hoek, T.L.V. Therapeutic hypothermia cardioprotection via Akt- and nitric oxide-mediated attenuation of mitochondrial oxidants. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2164–H2173. [Google Scholar]
- Poole, R.C.; Halestrap, A.P. Transport of lactate and other monocarboxylatesacross mammalian plasma membranes. Am. J. Physiol. 1993, 264, C761–C782. [Google Scholar]
- Hillered, L.; Vespa, P.M.; Hovda, D.A. Translational Neurochemical Research in Acute Human Brain Injury: The Current Status and Potential Future for Cerebral Microdialysis. J. Neurotrauma 2005, 22, 3–41. [Google Scholar] [CrossRef] [PubMed]
- Tisdall, M.M.; Smith, M. Cerebral microdialysis: Research technique or clinical tool. Br. J. Anaesth. 2006, 97, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Semenza, G.L. Hypoxia-inducible factors promote breast cancer stem cell specification and maintenance in response to hypoxia or cytotoxic chemotherapy. Adv. Cancer Res. 2019, 141, 175–212. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in Cellular Functions and Cell Death: Regulation and Biological Consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Chaui-Berlinck, J.G.; Monteiro, L.H.A.; Navas, C.A.; Bicudo, J.E.P.W. Temperature effects on energy metabolism: A dynamic system analysis. Proc. R. Soc. B Boil. Sci. 2002, 269, 15–19. [Google Scholar] [CrossRef]
- Geiser, F. Metabolic Rate and Body Temperature Reduction During Hibernation and Daily Torpor. Annu. Rev. Physiol. 2004, 66, 239–274. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, B. Evidence in support of a concept of reductive stress. Br. J. Nutr. 2002, 87, 93–94. [Google Scholar] [CrossRef]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef]
- Seki, T.; Yang, Y.; Sun, X.; Lim, S.; Xie, S.; Guo, Z.; Xiong, W.; Kuroda, M.; Sakaue, H.; Hosaka, K.; et al. Brown-fat-mediated tumour suppression by cold-altered global metabolism. Nature 2022, 608, 421–428. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms Underlying Acute Protection from Cardiac Ischemia-Reperfusion Injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Frei, B. Efficacy of Dietary Antioxidants to Prevent Oxidative Damage and Inhibit Chronic Disease. J. Nutr. 2004, 134, 3196S–3198S. [Google Scholar] [CrossRef] [PubMed]
- Porosk, R.; Terasmaa, A.; Mahlapuu, R.; Soomets, U.; Kilk, K. Metabolomics of the Wolfram Syndrome 1 Gene (Wfs1) Deficient Mice. J. Integr. Biol. 2017, 21, 721–732. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Beroud, C.; Demont, J.; Bouvier, R.; Schägger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Brière, J.-J.; Burnichon, N.; Rivière, J.; Vescovo, L.; Benit, P.; Giscos-Douriez, I.; De Reyniès, A.; Bertherat, J.; Badoual, C.; et al. The Warburg Effect Is Genetically Determined in Inherited Pheochromocytomas. PLoS ONE 2009, 4, e7094. [Google Scholar] [CrossRef]
- Moore, C.L.; Strasberg, P.M. Metabolic Reactions in the Nervous System; Springer: Berlin/Heidelberg, Germany, 1970; pp. 53–85. [Google Scholar]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free Radical. Bio. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef]
- Scandurra, F.M.; Gnaiger, E. Cell respiration under hypoxia: Facts and artefacts in mitochondrial oxygen kinetics. Adv Exp Med Biol. 2010, 662, 7–25. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eskla, K.-L.; Vellama, H.; Tarve, L.; Eichelmann, H.; Jagomäe, T.; Porosk, R.; Oja, V.; Rämma, H.; Peet, N.; Laisk, A.; et al. Hypothermia Alleviates Reductive Stress, a Root Cause of Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2022, 23, 10108. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231710108
Eskla K-L, Vellama H, Tarve L, Eichelmann H, Jagomäe T, Porosk R, Oja V, Rämma H, Peet N, Laisk A, et al. Hypothermia Alleviates Reductive Stress, a Root Cause of Ischemia Reperfusion Injury. International Journal of Molecular Sciences. 2022; 23(17):10108. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231710108
Chicago/Turabian StyleEskla, Kattri-Liis, Hans Vellama, Liisi Tarve, Hillar Eichelmann, Toomas Jagomäe, Rando Porosk, Vello Oja, Heikko Rämma, Nadežda Peet, Agu Laisk, and et al. 2022. "Hypothermia Alleviates Reductive Stress, a Root Cause of Ischemia Reperfusion Injury" International Journal of Molecular Sciences 23, no. 17: 10108. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231710108