
The Role of Insulin Receptor Substrate Proteins in Bronchopulmonary Dysplasia and Asthma: New Potential Perspectives

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Biological Role of Insulin Receptor Substrate Proteins in Human Tissue
3. Possible Roles of IRS in Pediatric Lung Diseases
3.1. Pediatric Asthma
3.2. Pediatric Asthma and IRS Signaling: A Forgotten Gap?
3.3. Bronchopulmonary Dysplasia
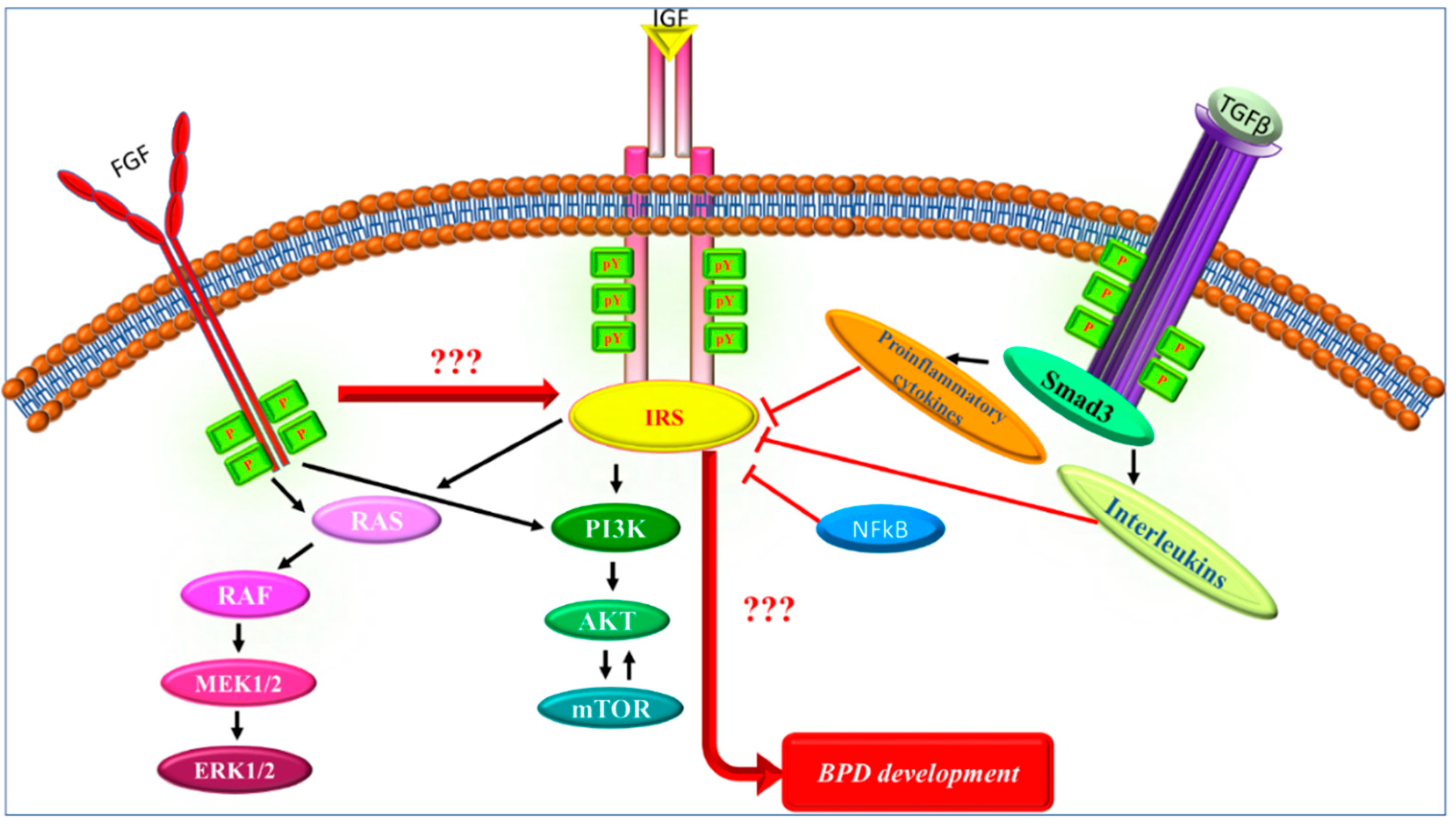
3.4. Aspects of IRS Signaling Possibly Involved in BPD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Cao, J.; Yee, D. Disrupting Insulin and IGF Receptor Function in Cancer. Int. J. Mol. Sci. 2021, 22, 555. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Kyohara, M.; Terauchi, Y.; Shirakawa, J. The Roles of the IGF Axis in the Regulation of the Metabolism: Interaction and Difference between Insulin Receptor Signaling and IGF-I Receptor Signaling. Int. J. Mol. Sci. 2021, 22, 6817. [Google Scholar] [CrossRef]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.Y.; Yan, Q.; Chen, W.; Forno, E.; Celedon, J.C. Serum insulin-like growth factor-1, asthma, and lung function among British adults. Ann. Allergy Asthma Immunol. 2021, 126, 284–291.e282. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, W.; Guo, Q.; Wang, Y.; Ma, L.; Zhang, X. Insulin-Like Growth Factor-1 Signaling in Lung Development and Inflammatory Lung Diseases. Biomed Res. Int. 2018, 2018, 6057589. [Google Scholar] [CrossRef]
- Gorgisen, G.; Karatas, U.; Ates, C.; Oksuz, M.; Gulacar, I.M. Association of IRS1 Gly972Arg and IRS2 Gly1057Asp polymorphisms with gastric cancer in Turkish subjects. Oncol. Lett. 2020, 20, 2016–2020. [Google Scholar] [CrossRef]
- Machado-Neto, J.A.; Fenerich, B.A.; Rodrigues Alves, A.P.N.; Fernandes, J.C.; Scopim-Ribeiro, R.; Coelho-Silva, J.L.; Traina, F. Insulin Substrate Receptor (IRS) proteins in normal and malignant hematopoiesis. Clinics 2018, 73, e566s. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signaling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Lavin, D.P.; White, M.F.; Brazil, D.P. IRS proteins and diabetic complications. Diabetologia 2016, 59, 2280–2291. [Google Scholar] [CrossRef]
- Gorgisen, G.; Hapil, F.Z.; Yilmaz, O.; Cetin, Z.; Pehlivanoglu, S.; Ozbudak, I.H.; Erdogan, A.; Ozes, O.N. Identification of novel mutations of Insulin Receptor Substrate 1 (IRS1) in tumor samples of non-small cell lung cancer (NSCLC): Implications for aberrant insulin signaling in development of cancer. Genet. Mol. Biol. 2019, 42, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, P.; Dorsey, N.J.; Li, J.; Qi, X.; Smith, E.P.; Yamaji-Kegan, K.; Keegan, A.D. The adaptor protein insulin receptor substrate 2 inhibits alternative macrophage activation and allergic lung inflammation. Sci. Signal. 2016, 9, ra63. [Google Scholar] [CrossRef] [PubMed]
- Hanke, S.; Mann, M. The phosphotyrosine interactome of the insulin receptor family and its substrates IRS-1 and IRS-2. Mol. Cell. Proteom. 2009, 8, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Voliovitch, H.; Schindler, D.G.; Hadari, Y.R.; Taylor, S.I.; Accili, D.; Zick, Y. Tyrosine phosphorylation of insulin receptor substrate-1 in vivo depends upon the presence of its pleckstrin homology region. J. Biol. Chem. 1995, 270, 18083–18087. [Google Scholar] [CrossRef] [PubMed]
- Yenush, L.; Makati, K.J.; Smith-Hall, J.; Ishibashi, O.; Myers, M.G., Jr.; White, M.F. The pleckstrin homology domain is the principal link between the insulin receptor and IRS-1. J. Biol. Chem. 1996, 271, 24300–24306. [Google Scholar] [CrossRef]
- Burks, D.J.; Pons, S.; Towery, H.; Smith-Hall, J.; Myers, M.G., Jr.; Yenush, L.; White, M.F. Heterologous pleckstrin homology domains do not couple IRS-1 to the insulin receptor. J. Biol. Chem. 1997, 272, 27716–27721. [Google Scholar] [CrossRef]
- Smith-Hall, J.; Pons, S.; Patti, M.E.; Burks, D.J.; Yenush, L.; Sun, X.J.; Kahn, C.R.; White, M.F. The 60 kDa insulin receptor substrate functions like an IRS protein (pp60IRS3) in adipose cells. Biochemistry 1997, 36, 8304–8310. [Google Scholar] [CrossRef]
- Lavan, B.E.; Fantin, V.R.; Chang, E.T.; Lane, W.S.; Keller, S.R.; Lienhard, G.E. A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J. Biol. Chem. 1997, 272, 21403–21407. [Google Scholar] [CrossRef]
- Cai, D.; Dhe-Paganon, S.; Melendez, P.A.; Lee, J.; Shoelson, S.E. Two new substrates in insulin signaling, IRS5/DOK4 and IRS6/DOK5. J. Biol. Chem. 2003, 278, 25323–25330. [Google Scholar] [CrossRef]
- Bjornholm, M.; He, A.R.; Attersand, A.; Lake, S.; Liu, S.C.; Lienhard, G.E.; Taylor, S.; Arner, P.; Zierath, J.R. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia 2002, 45, 1697–1702. [Google Scholar] [CrossRef]
- Favre, C.; Gerard, A.; Clauzier, E.; Pontarotti, P.; Olive, D.; Nunes, J.A. DOK4 and DOK5: New Dok-related genes expressed in human T cells. Genes Immun. 2003, 4, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Mardilovich, K.; Pankratz, S.L.; Shaw, L.M. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun. Signal. 2009, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Alex, J.M.; Bast, F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: Novel treatment strategies for cancer. Med. Oncol. 2014, 31, 805. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E413–E422. [Google Scholar] [CrossRef] [PubMed]
- Gorgisen, G.; Gulacar, I.M.; Ozes, O.N. The role of insulin receptor substrate (IRS) proteins in oncogenic transformation. Cell. Mol. Biol. 2017, 63, 1–5. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signaling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Deng, S.; Leong, H.C.; Datta, A.; Gopal, V.; Kumar, A.P.; Yap, C.T. PI3K/AKT Signaling Tips the Balance of Cytoskeletal Forces for Cancer Progression. Cancers 2022, 14, 1652. [Google Scholar] [CrossRef]
- Tanaka, S.; Ito, T.; Wands, J.R. Neoplastic transformation induced by insulin receptor substrate-1 overexpression requires an interaction with both Grb2 and Syp signaling molecules. J. Biol. Chem. 1996, 271, 14610–14616. [Google Scholar] [CrossRef]
- Hancer, N.J.; Qiu, W.; Cherella, C.; Li, Y.; Copps, K.D.; White, M.F. Insulin and metabolic stress stimulate multisite serine/threonine phosphorylation of insulin receptor substrate 1 and inhibit tyrosine phosphorylation. J. Biol. Chem. 2014, 289, 12467–12484. [Google Scholar] [CrossRef]
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304. [Google Scholar] [CrossRef]
- Tanti, J.F.; Jager, J. Cellular mechanisms of insulin resistance: Role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr. Opin. Pharmacol. 2009, 9, 753–762. [Google Scholar] [CrossRef]
- Ozes, O.N.; Akca, H.; Mayo, L.D.; Gustin, J.A.; Maehama, T.; Dixon, J.E.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc. Natl. Acad. Sci. USA 2001, 98, 4640–4645. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, K.; Haruta, T.; Tani, A.; Kawahara, J.; Usui, I.; Kobayashi, M. Roles of mTOR and JNK in serine phosphorylation, translocation, and degradation of IRS-1. Biochem. Biophys. Res. Commun. 2005, 335, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; He, L. IRS posttranslational modifications in regulating insulin signaling. J. Mol. Endocrinol. 2018, 60, R1–R8. [Google Scholar] [CrossRef] [PubMed]
- Toskala, E.; Kennedy, D.W. Asthma risk factors. Int. Forum. Allergy Rhinol. 2015, 5 (Suppl. 1), S11–S16. [Google Scholar] [CrossRef]
- Guibas, G.V.; Mathioudakis, A.G.; Tsoumani, M.; Tsabouri, S. Relationship of Allergy with Asthma: There Are More Than the Allergy “Eggs” in the Asthma “Basket”. Front. Pediatr. 2017, 5, 92. [Google Scholar] [CrossRef]
- Quirt, J.; Hildebrand, K.J.; Mazza, J.; Noya, F.; Kim, H. Asthma. Allergy Asthma Clin. Immunol. 2018, 14, 50. [Google Scholar] [CrossRef]
- Sandrock, C.E.; Norris, A. Infection in severe asthma exacerbations and critical asthma syndrome. Clin. Rev. Allergy Immunol. 2015, 48, 104–113. [Google Scholar] [CrossRef]
- Schivo, M.; Phan, C.; Louie, S.; Harper, R.W. Critical asthma syndrome in the ICU. Clin. Rev. Allergy Immunol. 2015, 48, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef]
- Wenzel, S. Severe asthma: From characteristics to phenotypes to endotypes. Clin. Exp. Allergy 2012, 42, 650–658. [Google Scholar] [CrossRef]
- Komlosi, Z.I.; van de Veen, W.; Kovacs, N.; Szucs, G.; Sokolowska, M.; O’Mahony, L.; Akdis, M.; Akdis, C.A. Cellular and molecular mechanisms of allergic asthma. Mol. Asp. Med. 2022, 85, 100995. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M. Asthma and allergic inflammation. Cell 2010, 140, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Mims, J.W. Asthma: Definitions and pathophysiology. Int. Forum. Allergy Rhinol. 2015, 5 (Suppl. 1), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, D.K.; Shao, Z. Pathogenesis of allergic airway inflammation. Curr. Allergy Asthma Rep. 2010, 10, 39–48. [Google Scholar] [CrossRef]
- Finn, P.W.; Bigby, T.D. Innate immunity and asthma. Proc. Am. Thorac. Soc. 2009, 6, 260–265. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; El-Gammal, A.I.; O’Byrne, P.M. Allergen-induced airway responses. Eur. Respir. J. 2015, 46, 819–831. [Google Scholar] [CrossRef]
- Kim, H.Y.; DeKruyff, R.H.; Umetsu, D.T. The many paths to asthma: Phenotype shaped by innate and adaptive immunity. Nat. Immunol. 2010, 11, 577–584. [Google Scholar] [CrossRef]
- Madore, A.M.; Laprise, C. Immunological and genetic aspects of asthma and allergy. J. Asthma Allergy 2010, 3, 107–121. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 1469–1485. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Heller, N.M.; Qi, X.; Junttila, I.S.; Shirey, K.A.; Vogel, S.N.; Paul, W.E.; Keegan, A.D. Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci. Signal. 2008, 1, ra17. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.M.; Gowda, N.; Fang, J.X.; Heller, N.M. Suppressor of Cytokine Signaling (SOCS)1 Regulates Interleukin-4 (IL-4)-activated Insulin Receptor Substrate (IRS)-2 Tyrosine Phosphorylation in Monocytes and Macrophages via the Proteasome. J. Biol. Chem. 2016, 291, 20574–20587. [Google Scholar] [CrossRef]
- Gour, N.; Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015, 75, 68–78. [Google Scholar] [CrossRef]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef]
- Sun, X.J.; Wang, L.M.; Zhang, Y.; Yenush, L.; Myers, M.G., Jr.; Glasheen, E.; Lane, W.S.; Pierce, J.H.; White, M.F. Role of IRS-2 in insulin and cytokine signaling. Nature 1995, 377, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Rabega, C.; Alexandrescu, R.; Rabega, M. Study of purified aldolase from Saccharomyces cerevisiae, using irradiated fructose-1,6-diphosphate. Rev. Ig Bacteriol. Virusol. Parazitol. Epidemiol. Pneumoftiziol. Bacteriol. Virusol. Parazitol. Epidemiol. 1976, 21, 37–41. [Google Scholar]
- Brisson-Lougarre, A.; Blum, C.J. Specific receptors for triiodothyronine in nuclei isolated from normal human polynuclear neutrophils. C. R. Acad. Sci. III 1985, 300, 287–292. [Google Scholar]
- Manohar, S.; Yu, Q.; Gygi, S.P.; King, R.W. The Insulin Receptor Adaptor IRS2 is an APC/C Substrate That Promotes Cell Cycle Protein Expression and a Robust Spindle Assembly Checkpoint. Mol. Cell. Proteom. 2020, 19, 1450–1467. [Google Scholar] [CrossRef]
- Nakahara, M.; Ito, H.; Skinner, J.T.; Lin, Q.; Tamosiuniene, R.; Nicolls, M.R.; Keegan, A.D.; Johns, R.A.; Yamaji-Kegan, K. The inflammatory role of dysregulated IRS2 in pulmonary vascular remodeling under hypoxic conditions. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L416–L428. [Google Scholar] [CrossRef]
- Keegan, A.D.; Zamorano, J.; Keselman, A.; Heller, N.M. IL-4 and IL-13 Receptor Signaling From 4PS to Insulin Receptor Substrate 2: There and Back Again, a Historical View. Front. Immunol. 2018, 9, 1037. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.J.; Fang, X.; Gowda, N.M.; Thompson, J.J.; Heller, N.M. The TORC1-activated Proteins, p70S6K and GRB10, Regulate IL-4 Signaling and M2 Macrophage Polarization by Modulating Phosphorylation of Insulin Receptor Substrate-2. J. Biol. Chem. 2016, 291, 24922–24930. [Google Scholar] [CrossRef] [Green Version]
- Karo-Atar, D.; Bitton, A.; Benhar, I.; Munitz, A. Therapeutic Targeting of the Interleukin-4/Interleukin-13 Signaling Pathway: In Allergy and Beyond. BioDrugs 2018, 32, 201–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.J.; Wang, S.H.; Wu, C.C.; Su, Y.A.; Chiang, C.Y.; Lai, C.H.; Wang, T.H.; Cheng, T.L.; Kuo, J.Y.; Hsu, T.C.; et al. IL-4 and IL-13 Promote Proliferation of Mammary Epithelial Cells through STAT6 and IRS-1. Int. J. Mol. Sci. 2021, 22, 12008. [Google Scholar] [CrossRef]
- Keegan, A.D.; Nelms, K.; White, M.; Wang, L.M.; Pierce, J.H.; Paul, W.E. An IL-4 receptor region containing an insulin receptor motif is important for IL-4-mediated IRS-1 phosphorylation and cell growth. Cell 1994, 76, 811–820. [Google Scholar] [CrossRef]
- White, S.R.; Martin, L.D.; Abe, M.K.; Marroquin, B.A.; Stern, R.; Fu, X. Insulin receptor substrate-1/2 mediates IL-4-induced migration of human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L164–L173. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Tanaka, T.; Shi, W.; Matsumoto, M.; Minami, M.; Kashiwamura, S.; Nakanishi, K.; Yoshida, N.; Kishimoto, T.; Akira, S. Essential role of Stat6 in IL-4 signaling. Nature 1996, 380, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, L.; Hao, S.; Liu, Z.; Ding, S.; Zhang, W.; Yang, X.; Li, S. Activation of the IL-4/STAT6 Signaling Pathway Promotes Lung Cancer Progression by Increasing M2 Myeloid Cells. Front. Immunol. 2019, 10, 2638. [Google Scholar] [CrossRef]
- Northway, W.H., Jr.; Rosan, R.C.; Porter, D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N. Engl. J. Med. 1967, 276, 357–368. [Google Scholar] [CrossRef]
- Tracy, M.K.; Berkelhamer, S.K. Bronchopulmonary Dysplasia and Pulmonary Outcomes of Prematurity. Pediatr. Ann. 2019, 48, e148–e153. [Google Scholar] [CrossRef]
- Bhandari, A.; Bhandari, V. Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics 2009, 123, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.J. The new BPD: An arrest of lung development. Pediatr. Res. 1999, 46, 641–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobe, A.H.; Bancalari, E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 163, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Davidson, L.M.; Berkelhamer, S.K. Bronchopulmonary Dysplasia: Chronic Lung Disease of Infancy and Long-Term Pulmonary Outcomes. J. Clin. Med. 2017, 6, 4. [Google Scholar] [CrossRef]
- Thebaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Warner, B.B.; Stuart, L.A.; Papes, R.A.; Wispe, J.R. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am. J. Physiol. 1998, 275, L110–L117. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Chong, L.; Bellusci, S. Fgf10/Fgfr2b Signaling Orchestrates the Symphony of Molecular, Cellular, and Physical Processes Required for Harmonious Airway Branching Morphogenesis. Front. Cell Dev. Biol. 2020, 8, 620667. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, S.; Chao, C.M.; Guenther, S.; Glaser, L.; Gersmann, L.; Michel, G.; Kraut, S.; Goth, K.; Koepke, J.; Heiner, M.; et al. FGF10 triggers de novo alveologenesis in a BPD model: Impact on the resident mesenchymal niche cells. Stem. Cells 2022, 40, 605–617. [Google Scholar] [CrossRef]
- Oak, P.; Hilgendorff, A. The BPD trio? Interaction of dysregulated PDGF, VEGF, and TGF signaling in neonatal chronic lung disease. Mol. Cell. Pediatr. 2017, 4, 11. [Google Scholar] [CrossRef]
- Huang, S.S.; Leal, S.M.; Chen, C.L.; Liu, I.H.; Huang, J.S. Cellular growth inhibition by TGF-beta1 involves IRS proteins. FEBS Lett. 2004, 565, 117–121. [Google Scholar] [CrossRef]
- Yoneyama, Y.; Lanzerstorfer, P.; Niwa, H.; Umehara, T.; Shibano, T.; Yokoyama, S.; Chida, K.; Weghuber, J.; Hakuno, F.; Takahashi, S.I. IRS-1 acts as an endocytic regulator of IGF-I receptor to facilitate sustained IGF signaling. Elife 2018, 7, e32893. [Google Scholar] [CrossRef] [PubMed]
- Rabiee, A.; Kruger, M.; Ardenkjaer-Larsen, J.; Kahn, C.R.; Emanuelli, B. Distinct signaling properties of insulin receptor substrate (IRS)-1 and IRS-2 in mediating insulin/IGF-1 action. Cell. Signal. 2018, 47, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.L.; Ryan, R.M. Bronchopulmonary dysplasia: New becomes old again! Pediatr. Res. 2017, 81, 210–213. [Google Scholar] [CrossRef]
- Capoluongo, E.; Ameglio, F.; Zuppi, C. Insulin-like growth factor-I and complications of prematurity: A focus on bronchopulmonary dysplasia. Clin. Chem. Lab. Med. 2008, 46, 1061–1066. [Google Scholar] [CrossRef]
- Chetty, A.; Nielsen, H.C. Regulation of cell proliferation by insulin-like growth factor 1 in hyperoxia-exposed neonatal rat lung. Mol. Genet. Metab. 2002, 75, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Belcastro, R.; Lopez, L.; Li, J.; Masood, A.; Tanswell, A.K. Chronic lung injury in the neonatal rat: Up-regulation of TGFbeta1 and nitration of IGF-R1 by peroxynitrite as likely contributors to impaired alveologenesis. Free Radic. Biol. Med. 2015, 80, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, K.; Epaud, R.; Holzenberger, M.; Bonora, M.; Flejou, J.F.; Puard, J.; Clement, A.; Henrion-Caude, A. Deficiency in type 1 insulin-like growth factor receptor in mice protects against oxygen-induced lung injury. Respir. Res. 2005, 6, 31. [Google Scholar] [CrossRef]
- Kheirollahi, V.; Khadim, A.; Kiliaris, G.; Korfei, M.; Barroso, M.M.; Alexopoulos, I.; Vazquez-Armendariz, A.I.; Wygrecka, M.; Ruppert, C.; Guenther, A.; et al. Transcriptional Profiling of Insulin-like Growth Factor Signaling Components in Embryonic Lung Development and Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 1973. [Google Scholar] [CrossRef]
- Lofqvist, C.; Hellgren, G.; Niklasson, A.; Engstrom, E.; Ley, D.; Hansen-Pupp, I.; Consortium, W. Low postnatal serum IGF-I levels are associated with bronchopulmonary dysplasia (BPD). Acta Paediatr. 2012, 101, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Chetty, A.; Andersson, S.; Lassus, P.; Nielsen, H.C. Insulin-like growth factor-1 (IGF-1) and IGF-1 receptor (IGF-1R) expression in human lung in RDS and BPD. Pediatr. Pulmonol. 2004, 37, 128–136. [Google Scholar] [CrossRef]
- Capoluongo, E.; Vento, G.; Ameglio, F.; Lulli, P.; Matassa, P.G.; Carrozza, C.; Santini, S.A.; Antenucci, M.; Castagnola, M.; Giardina, B.; et al. Increased levels of IGF-1 and beta2-microglobulin in epithelial lining fluid of preterm newborns developing chronic lung disease. effects of rhG-CSF. Int. J. Immunopathol. Pharmacol. 2006, 19, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Luan, X.; Li, H.; Jin, Z. Insulin-like growth factor-1: A potential target for bronchopulmonary dysplasia treatment (Review). Exp. Ther. Med. 2022, 23, 191. [Google Scholar] [CrossRef] [PubMed]
- Kuebler, W.M.; Uhlig, U.; Goldmann, T.; Schael, G.; Kerem, A.; Exner, K.; Martin, C.; Vollmer, E.; Uhlig, S. Stretch activates nitric oxide production in pulmonary vascular endothelial cells in situ. Am. J. Respir. Crit. Care Med. 2003, 168, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, U.; Fehrenbach, H.; Lachmann, R.A.; Goldmann, T.; Lachmann, B.; Vollmer, E.; Uhlig, S. Phosphoinositide 3-OH kinase inhibition prevents ventilation-induced lung cell activation. Am. J. Respir. Crit. Care Med. 2004, 169, 201–208. [Google Scholar] [CrossRef]
- Benjamin, J.T.; Carver, B.J.; PLoSa, E.J.; Yamamoto, Y.; Miller, J.D.; Liu, J.H.; van der Meer, R.; Blackwell, T.S.; Prince, L.S. NF-kappaB activation limits airway branching through inhibition of Sp1-mediated fibroblast growth factor-10 expression. J. Immunol. 2010, 185, 4896–4903. [Google Scholar] [CrossRef]
- Carver, B.J.; PLoSa, E.J.; Stinnett, A.M.; Blackwell, T.S.; Prince, L.S. Interactions between NF-kappaB and SP3 connect inflammatory signaling with reduced FGF-10 expression. J. Biol. Chem. 2013, 288, 15318–15325. [Google Scholar] [CrossRef]
- Wu, D.; Liang, M.; Dang, H.; Fang, F.; Xu, F.; Liu, C. Hydrogen protects against hyperoxia-induced apoptosis in type II alveolar epithelial cells via activation of PI3K/Akt/Foxo3a signaling pathway. Biochem. Biophys. Res. Commun. 2018, 495, 1620–1627. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef]
- Wang, S.H.; Li, L.H.; Zou, D.M.; Zheng, X.M.; Deng, J. Roles of the mammalian target of rapamycin (mTOR) signaling pathway in the repair of hyperoxia-induced acute lung injury. Adv. Clin. Exp. Med. 2020, 29, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.J.; Sowers, A.; Thetford, A.; McKay-Corkum, G.; Chung, S.I.; Mitchell, J.B.; Citrin, D.E. Mammalian Target of Rapamycin Inhibition With Rapamycin Mitigates Radiation-Induced Pulmonary Fibrosis in a Murine Model. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ma, Q.; Ma, X.; Zhang, Z.; Liu, N.; Wang, M. Role of mammalian target of rapamycin signaling in autophagy and the neurodegenerative process using a senescence accelerated mouse-prone 8 model. Exp. Ther. Med. 2017, 14, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Porzionato, A.; Sfriso, M.M.; Mazzatenta, A.; Macchi, V.; De Caro, R.; Di Giulio, C. Effects of hyperoxic exposure on signal transduction pathways in the lung. Respir. Physiol. Neurobiol. 2015, 209, 106–114. [Google Scholar] [CrossRef]
- Rubinfeld, H.; Seger, R. The ERK cascade: A prototype of MAPK signaling. Mol. Biotechnol. 2005, 31, 151–174. [Google Scholar] [CrossRef]
- El Agha, E.; Bellusci, S. Walking along the Fibroblast Growth Factor 10 Route: A Key Pathway to Understand the Control and Regulation of Epithelial and Mesenchymal Cell-Lineage Formation during Lung Development and Repair after Injury. Scientifica 2014, 2014, 538379. [Google Scholar] [CrossRef]
- Bellusci, S.; Grindley, J.; Emoto, H.; Itoh, N.; Hogan, B.L. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 1997, 124, 4867–4878. [Google Scholar] [CrossRef]
- Gupte, V.V.; Ramasamy, S.K.; Reddy, R.; Lee, J.; Weinreb, P.H.; Violette, S.M.; Guenther, A.; Warburton, D.; Driscoll, B.; Minoo, P.; et al. Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 424–436. [Google Scholar] [CrossRef]
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; De Langhe, S.P. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Investig. 2011, 121, 4409–4419. [Google Scholar] [CrossRef]
- Alejandre-Alcazar, M.A.; Michiels-Corsten, M.; Vicencio, A.G.; Reiss, I.; Ryu, J.; de Krijger, R.R.; Haddad, G.G.; Tibboel, D.; Seeger, W.; Eickelberg, O.; et al. TGF-beta signaling is dynamically regulated during the alveolarization of rodent and human lungs. Dev. Dyn. 2008, 237, 259–269. [Google Scholar] [CrossRef]
- Chao, C.M.; Chong, L.; Chu, X.; Shrestha, A.; Behnke, J.; Ehrhardt, H.; Zhang, J.; Chen, C.; Bellusci, S. Targeting Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension (BPD-PH): Potential Role of the FGF Signaling Pathway in the Development of the Pulmonary Vascular System. Cells 2020, 9, 1875. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Walker, D.J.; Cwiklinski, E.; Tait, C.; Tee, A.R.; Land, S.C. Control of HIF-1α and vascular signaling in fetal lung involves cross talk between mTORC1 and the FGF-10/FGFR2b/Spry2 airway branching periodicity clock. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L455–L471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Alam, D.; El Agha, E.; Sakurai, R.; Kheirollahi, V.; Moiseenko, A.; Danopoulos, S.; Shrestha, A.; Schmoldt, C.; Quantius, J.; Herold, S.; et al. Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung development. Development 2015, 142, 4139–4150. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.K.; Jankowska, L.; Prisco, M.; Xu, S.; Guvakova, M.A.; Surmacz, E. Differential roles of IRS-1 and SHC signaling pathways in breast cancer cells. Int. J. Cancer 1997, 72, 828–834. [Google Scholar] [CrossRef]
- Shi, Y.; Ma, Z.; Cheng, Q.; Wu, Y.; Parris, A.B.; Kong, L.; Yang, X. FGFR1 overexpression renders breast cancer cells resistant to metformin through activation of IRS1/ERK signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118877. [Google Scholar] [CrossRef] [PubMed]
- Dailey, L.; Laplantine, E.; Priore, R.; Basilico, C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J. Cell Biol. 2003, 161, 1053–1066. [Google Scholar] [CrossRef]
- Lassarre, C.; Ricort, J.M. Growth factor-specific regulation of insulin receptor substrate-1 expression in MCF-7 breast carcinoma cells: Effects on the insulin-like growth factor signaling pathway. Endocrinology 2003, 144, 4811–4819. [Google Scholar] [CrossRef]
- Manzano-Nunez, F.; Arambul-Anthony, M.J.; Galan Albinana, A.; Leal Tassias, A.; Acosta Umanzor, C.; Borreda Gasco, I.; Herrera, A.; Forteza Vila, J.; Burks, D.J.; Noon, L.A. Insulin resistance disrupts epithelial repair and niche-progenitor Fgf signaling during chronic liver injury. PLoS Biol. 2019, 17, e2006972. [Google Scholar] [CrossRef]
- Alejandre-Alcazar, M.A.; Kwapiszewska, G.; Reiss, I.; Amarie, O.V.; Marsh, L.M.; Sevilla-Perez, J.; Wygrecka, M.; Eul, B.; Kobrich, S.; Hesse, M.; et al. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L537–L549. [Google Scholar] [CrossRef]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Gauldie, J.; Galt, T.; Bonniaud, P.; Robbins, C.; Kelly, M.; Warburton, D. Transfer of the active form of transforming growth factor-beta 1 gene to newborn rat lung induces changes consistent with bronchopulmonary dysplasia. Am. J. Pathol. 2003, 163, 2575–2584. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Syed, M.A.; Boddupalli, C.S.; Dhodapkar, M.V.; Homer, R.J.; Minoo, P.; Bhandari, V. Conditional overexpression of TGFbeta1 promotes pulmonary inflammation, apoptosis and mortality via TGFbetaR2 in the developing mouse lung. Respir. Res. 2015, 16, 4. [Google Scholar] [CrossRef] [Green Version]
- Warburton, D.; Bellusci, S.; De Langhe, S.; Del Moral, P.M.; Fleury, V.; Mailleux, A.; Tefft, D.; Unbekandt, M.; Wang, K.; Shi, W. Molecular mechanisms of early lung specification and branching morphogenesis. Pediatr. Res. 2005, 57, 26R–37R. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorff, A.; Parai, K.; Ertsey, R.; Juliana Rey-Parra, G.; Thebaud, B.; Tamosiuniene, R.; Jain, N.; Navarro, E.F.; Starcher, B.C.; Nicolls, M.R.; et al. Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L215–L227. [Google Scholar] [CrossRef] [PubMed]
- Mokres, L.M.; Parai, K.; Hilgendorff, A.; Ertsey, R.; Alvira, C.M.; Rabinovitch, M.; Bland, R.D. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L23–L35. [Google Scholar] [CrossRef] [PubMed]
- Kompass, K.S.; Deslee, G.; Moore, C.; McCurnin, D.; Pierce, R.A. Highly conserved transcriptional responses to mechanical ventilation of the lung. Physiol. Genom. 2010, 42, 384–396. [Google Scholar] [CrossRef]
- Speer, C.P. Pulmonary inflammation and bronchopulmonary dysplasia. J. Perinatol. 2006, 26 (Suppl. 1), S57–S62. [Google Scholar] [CrossRef]
- Ballabh, P.; Simm, M.; Kumari, J.; Krauss, A.N.; Jain, A.; Califano, C.; Lesser, M.L.; Cunningham-Rundles, S. Neutrophil and monocyte adhesion molecules in bronchopulmonary dysplasia, and effects of corticosteroids. Arch. Dis. Child. Fetal Neonatal Ed. 2004, 89, F76–F83. [Google Scholar] [CrossRef]
- Ogden, B.E.; Murphy, S.; Saunders, G.C.; Johnson, J.D. Lung lavage of newborns with respiratory distress syndrome. Prolonged neutrophil influx is associated with bronchopulmonary dysplasia. Chest 1983, 83, 31S–33S. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Smale, G.; Pencev, D. Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils. J. Cell. Physiol. 1989, 140, 396–402. [Google Scholar] [CrossRef]
- Schultz, C.; Tautz, J.; Reiss, I.; Moller, J.C. Prolonged mechanical ventilation induces pulmonary inflammation in preterm infants. Biol. Neonate 2003, 84, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Merritt, T.A.; Deming, D.D.; Boynton, B.R. The ‘new’ bronchopulmonary dysplasia: Challenges and commentary. Semin. Fetal Neonatal Med. 2009, 14, 345–357. [Google Scholar] [CrossRef]
- Carlton, D.P.; Albertine, K.H.; Cho, S.C.; Lont, M.; Bland, R.D. Role of neutrophils in lung vascular injury and edema after premature birth in lambs. J. Appl. Physiol. 1997, 83, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Jaarsma, A.S.; Braaksma, M.A.; Geven, W.B.; van Oeveren, W.; Bambang Oetomo, S. Activation of the inflammatory reaction within minutes after birth in ventilated preterm lambs with neonatal respiratory distress syndrome. Biol. Neonate 2004, 86, 1–5. [Google Scholar] [CrossRef]
- Kotecha, S.; Mildner, R.J.; Prince, L.R.; Vyas, J.R.; Currie, A.E.; Lawson, R.A.; Whyte, M.K. The role of neutrophil apoptosis in the resolution of acute lung injury in newborn infants. Thorax 2003, 58, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Leal, S.M.; Chen, C.L.; Liu, I.H.; Huang, J.S. Identification of insulin receptor substrate proteins as key molecules for the TbetaR-V/LRP-1-mediated growth inhibitory signaling cascade in epithelial and myeloid cells. FASEB J. 2004, 18, 1719–1721. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, D.M.; Wang, C.M.; Hu, Y.; Liu, A.H.; Zhang, Y.L.; Sun, B.; Song, J.G. Insulin receptor substrate-1 suppresses transforming growth factor-beta1-mediated epithelial-mesenchymal transition. Cancer Res. 2009, 69, 7180–7187. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.L.; Agarwal, E.; Chowdhury, S.; Luo, J.; Brattain, M.G.; Black, J.D.; Wang, J. TGFbeta/Smad3 regulates proliferation and apoptosis through IRS-1 inhibition in colon cancer cells. PLoS ONE 2017, 12, e0176096. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorgisen, G.; Aydin, M.; Mboma, O.; Gökyildirim, M.Y.; Chao, C.-M. The Role of Insulin Receptor Substrate Proteins in Bronchopulmonary Dysplasia and Asthma: New Potential Perspectives. Int. J. Mol. Sci. 2022, 23, 10113. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231710113
Gorgisen G, Aydin M, Mboma O, Gökyildirim MY, Chao C-M. The Role of Insulin Receptor Substrate Proteins in Bronchopulmonary Dysplasia and Asthma: New Potential Perspectives. International Journal of Molecular Sciences. 2022; 23(17):10113. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231710113
Chicago/Turabian StyleGorgisen, Gokhan, Malik Aydin, Olivier Mboma, Mira Y. Gökyildirim, and Cho-Ming Chao. 2022. "The Role of Insulin Receptor Substrate Proteins in Bronchopulmonary Dysplasia and Asthma: New Potential Perspectives" International Journal of Molecular Sciences 23, no. 17: 10113. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231710113