
Advances of Epigenetic Biomarkers and Epigenome Editing for Early Diagnosis in Breast Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
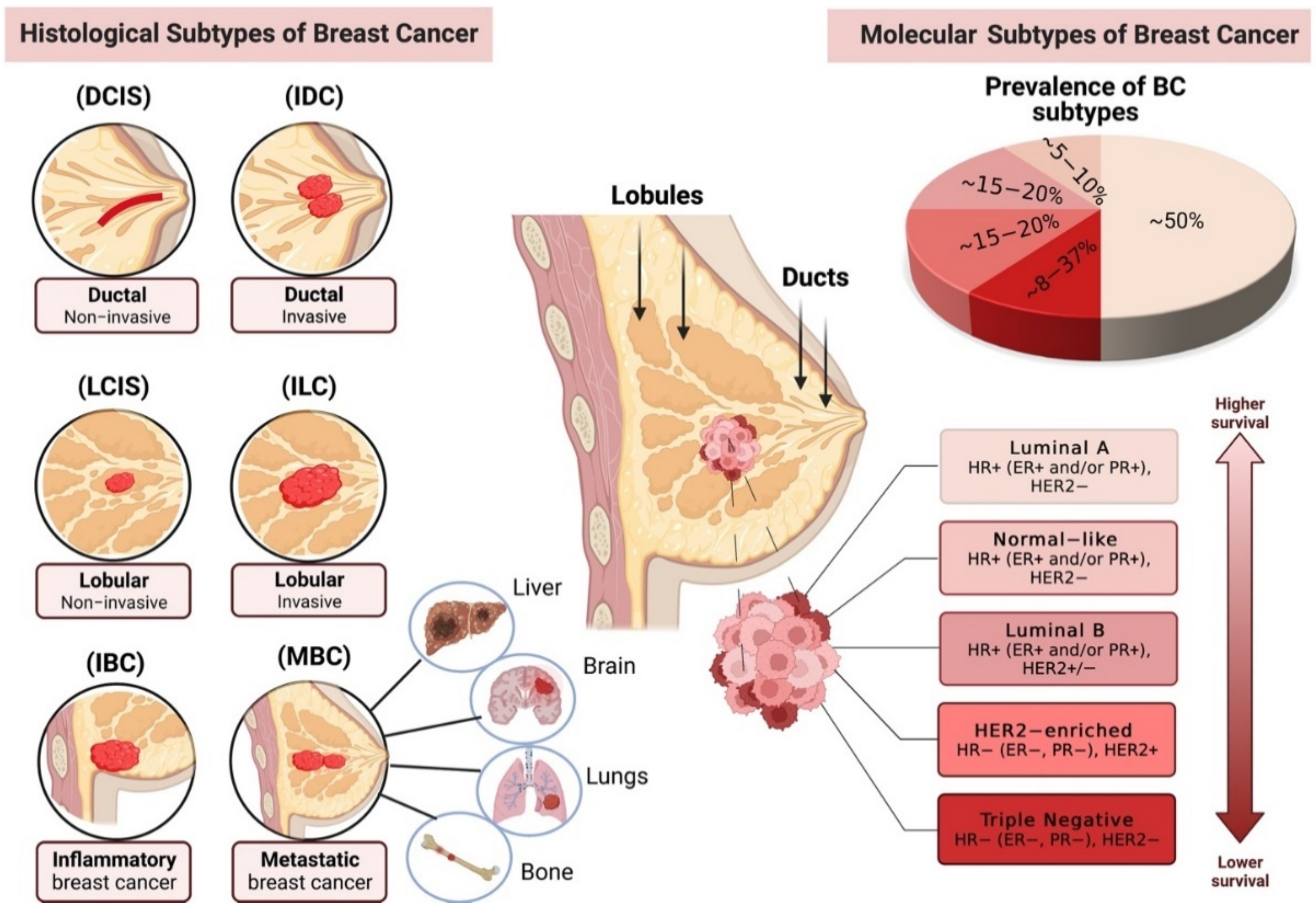
:1. Breast Cancer Pathogenesis and Subtypes (Histological and Molecular)

2. Epigenetic Modifications in Breast Cancer
2.1. DNA Methylation in Breast Cancer
2.2. Histone Modifications in Breast Cancer
2.3. Non-Coding RNAs (ncRNAs) in Breast Cancer
2.4. Non-Canonical Epigenetic Modification in Breast Cancer
3. Future Perspectives and Novel Strategies for Breast Cancer
3.1. Novel Diagnostic and Prognostic Epigenetic Biomarkers for BC Detection
3.2. Epigenetic-Modifying Drugs for Therapeutic Intervention in BC
3.3. Epigenetic Editing as a Valuable Tool for Rewriting or Erasing Aberrant Epigenetic Marks in BC
3.4. Biorecognition Engineering and Partitioning of Anticancer Drugs to Improve BC Therapeutics
3.5. Nanoparticle-Based Drug Delivery to Overcome Drug Resistance in BC Treatment
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Olopade, O.I.; Grushko, T.A.; Nanda, R.; Huo, D. Advances in Breast Cancer: Pathways to Personalized Medicine. Clin. Cancer Res. 2008, 14, 7988–7999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makki, J.J. Diversity of breast carcinoma: Histological subtypes and clinical relevance. Clin. Med. Insights Pathol. 2015, 8, CPath-S31563. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.; Gupta, S. The role of histone deacetylases in prostate cancer. Epigenetics 2008, 3, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Vizoso, M.; Ferreira, H.J.; Lopez-Serra, P.; Carmona, F.J.; Martínez-Cardús, A.; Girotti, M.R.; Villanueva, A.; Guil, S.; Moutinho, C.; Liz, J.; et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nat. Med. 2015, 21, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Dillekås, H.; Demicheli, R.; Ardoino, I.; Jensen, S.A.H.; Biganzoli, E.; Straume, O. The recurrence pattern following delayed breast reconstruction after mastectomy for breast cancer suggests a systemic effect of surgery on occult dormant micrometastases. Breast Cancer Res. Treat. 2016, 158, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Gudaviciene, D.; Steponaviciene, L.; Meskauskas, R.; Smailyte, G.; Aleknavicius, E. Rare types of breast carcinoma. Open Med. 2014, 10, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Masood, S. Breast cancer subtypes: Morphologic and biologic characterization. Women’s Health 2016, 12, 103–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappikeri, V.K.S.; Kriplani, A.M. Bilateral synchronous carcinoma breast-a rare case presentation. SpringerPlus 2015, 4, 193. [Google Scholar] [CrossRef] [Green Version]
- Pegram, M.D.; Zong, Y.; Yam, C.; Goetz, M.P.; Moulder, S.L. Innovative Strategies: Targeting Subtypes in Metastatic Breast Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Urbano, N.; Bonfiglio, R.; Duggento, A.; Toschi, N.; Schillaci, O.; Bonanno, E. Novel insights into breast cancer progression and metastasis: A multidisciplinary opportunity to transition from biology to clinical oncology. Biochim. Biophys. Acta 2019, 1872, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Sher, G.; Salman, N.A.; Khan, A.Q.; Prabhu, K.S.; Raza, A.; Kulinski, M.; Dermime, S.; Haris, M.; Junejo, K.; Uddin, S. Epigenetic and breast cancer therapy: Promising diagnostic and therapeutic applications. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2022; Volume 83, pp. 152–165. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.; Paish, E.C.; Powe, D.G.; Gee, J.; Nicholson, R.I.; Lee, A.H.; Robertson, J.F.; Ellis, I. Biologic and Clinical Characteristics of Breast Cancer With Single Hormone Receptor–Positive Phenotype. J. Clin. Oncol. 2007, 25, 4772–4778. [Google Scholar] [CrossRef]
- Zubair, M.; Wang, S.; Ali, N. Advanced Approaches to Breast Cancer Classification and Diagnosis. Front. Pharmacol. 2021, 11, 632079. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Tao, Y.; Young, O.; White, S.; Proia, A.D.; Murray, J.; Renshaw, L.; Faratian, D.; Thomas, J.; Dowsett, M.; et al. Estrogen-Independent Proliferation Is Present in Estrogen-Receptor HER2-Positive Primary Breast Cancer After Neoadjuvant Letrozole. J. Clin. Oncol. 2006, 24, 3019–3025. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Press, M.F. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 44, 707–712. [Google Scholar] [CrossRef]
- Godoy-Ortiz, A.; Sanchez-Muñoz, A.; Parrado, M.R.C.; Álvarez, M.; Ribelles, N.; Dominguez, A.R.; Alba, E. Deciphering HER2 Breast Cancer Disease: Biological and Clinical Implications. Front. Oncol. 2019, 9, 1124. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Jiang, T.; Shi, W.; Wali, V.B.; Pongor, L.S.; Li, C.; Lau, R.; Győrffy, B.; Lifton, R.P.; Symmans, W.F.; Pusztai, L.; et al. Predictors of Chemosensitivity in Triple Negative Breast Cancer: An Integrated Genomic Analysis. PLoS Med. 2016, 13, e1002193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantino, P.; Corti, C.; Schmid, P.; Cortes, J.; Mittendorf, E.A.; Rugo, H.; Tolaney, S.M.; Bianchini, G.; Andrè, F.; Curigliano, G. Immunotherapy for early triple negative breast cancer: Research agenda for the next decade. npj Breast Cancer 2022, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Szymiczek, A.; Lone, A.; Akbari, M.R. Molecular intrinsic versus clinical subtyping in breast cancer: A comprehensive review. Clin. Genet. 2021, 99, 613–637. [Google Scholar] [CrossRef] [PubMed]
- Vallard, A.; Magné, N.; Guy, J.B.; Espenel, S.; Rancoule, C.; Diao, P.; Chargari, C. Is breast-conserving therapy adequate in BRCA 1/2 mutation carriers? The radiation oncologist’s point of view. Br. J. Radiol. 2019, 92, 20170657. [Google Scholar] [CrossRef]
- Dobrovic, A.; Simpfendorfer, D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997, 57, 3347–3350. [Google Scholar]
- Tian, S.; Fu, L.; Zhang, J.; Xu, J.; Yuan, L.; Qin, J.; Zhang, W. Identification of a DNA Methylation-Driven Genes-Based Prognostic Model and Drug Targets in Breast Cancer: In silico Screening of Therapeutic Compounds and in vitro Characterization. Front. Immunol. 2021, 12, 4385. [Google Scholar] [CrossRef]
- Shukla, S.; Penta, D.; Mondal, P.; Meeran, S.M. Epigenetics of Breast Cancer: Clinical Status of Epi-drugs and Phytochemicals. In Breast Cancer Metastasis and Drug Resistance; Springer: Cham, Switzerland, 2019; pp. 293–310. [Google Scholar] [CrossRef]
- Zhuang, J.; Huo, Q.; Yang, F.; Xie, N. Perspectives on the Role of Histone Modification in Breast Cancer Progression and the Advanced Technological Tools to Study Epigenetic Determinants of Metastasis. Front. Genet. 2020, 11, 603552. [Google Scholar] [CrossRef]
- Elliott, R.A.; McDowell, J.; Marriott, J.L.; Calandra, A.; Duncan, G. A Pharmacy Preregistration Course Using Online Teaching and Learning Methods. Am. J. Pharm. Educ. 2009, 73, 77. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: An intimate affair. Am. J. Cancer Res. 2020, 10, 1954. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Brane, A.C.; Tollefsbol, T.O. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells 2019, 8, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, M.; Salta, S.; Henrique, R.; Jerónimo, C. Decoding the usefulness of non-coding RNAs as breast cancer markers. J. Transl. Med. 2016, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Faldoni, F.L.C.; Rainho, C.A.; Rogatto, S.R. Epigenetics in Inflammatory Breast Cancer: Biological Features and Therapeutic Perspectives. Cells 2020, 9, 1164. [Google Scholar] [CrossRef]
- Tiffon, C. The impact of nutrition and environmental epigenetics on human health and disease. Int. J. Mol. Sci. 2018, 9, 3425. [Google Scholar] [CrossRef] [Green Version]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Fowden, A.L.; Forhead, A.J. Hormones as epigenetic signals in developmental programming. Exp. Physiol. 2009, 94, 607–625. [Google Scholar] [CrossRef]
- Olmedo-Suárez, M.; Ramírez-Díaz, I.; Pérez-González, A.; Molina-Herrera, A.; Coral-García, M.; Lobato, S.; Sarvari, P.; Barreto, G.; Rubio, K. Epigenetic Regulation in Exposome-Induced Tumorigenesis: Emerging Roles of ncRNAs. Biomolecules 2022, 12, 513. [Google Scholar] [CrossRef]
- Loison, L. Epigenetic inheritance and evolution: A historian’s perspective. Philo. Trans. R. Soc. B 2021, 376, 20200120. [Google Scholar] [CrossRef]
- Almouzni, G.; Cedar, H. Maintenance of epigenetic information. Cold Spring Harb. Perspect. Biol. 2016, 8, a019372. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of Development and Genetic Assimilation of Acquired Characters. Nature 1959, 183, 1654–1655. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The epigenotype. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Günther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.-M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, T.M.; Søreide, K. Cancer epigenetics in solid organ tumours: A primer for surgical oncologists. Eur. J. Surg. Oncol. 2019, 45, 736–746. [Google Scholar] [CrossRef]
- Chen, J.F.; Yan, Q. The roles of epigenetics in cancer progression and metastasis. Biochem. J. 2021, 478, 3373–3393. [Google Scholar] [CrossRef]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Mirza, S.; Sharma, G.; Parshad, R.; Gupta, S.D.; Pandya, P.; Ralhan, R. Expression of DNA Methyltransferases in Breast Cancer Patients and to Analyze the Effect of Natural Compounds on DNA Methyltransferases and Associated Proteins. J. Breast Cancer 2013, 16, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Cheung, H.H.; Lee, T.-L.; Rennert, O.M.; Chan, W.Y. DNA methylation of cancer genome. Birth Defects Res. Part C Embryo Today Rev. 2009, 87, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation of ras oncogenes in primary human cancers. Biochem. Biophys. Res. Commun. 1983, 111, 47–54. [Google Scholar] [CrossRef]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Severi, G.; Southey, M.C.; English, D.; Jung, C.-H.; Lonie, A.; McLean, C.; Tsimiklis, H.; Hopper, J.L.; Giles, G.; Baglietto, L. Epigenome-wide methylation in DNA from peripheral blood as a marker of risk for breast cancer. Breast Cancer Res. Treat. 2014, 148, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Cruzata, L.; Wu, H.-C.; Liao, Y.; Santella, R.M.; Terry, M.B. Differences in DNA methylation by extent of breast cancer family history in unaffected women. Epigenetics 2014, 9, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Veldhoven, K.; Polidoro, S.; Baglietto, L.; Severi, G.; Sacerdote, C.; Panico, S.; Vineis, P. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin. Epigenetics 2015, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Garcia-Closas, M.; Orr, N.; Fletcher, O.; Jones, M.; Ashworth, A.; Flanagan, J.M. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012, 72, 2304–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, T.; Yamamoto, N.; Taguchi, T.; Tamaki, Y.; Noguchi, S. BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast cancer with BRCA1 promoter methylation. Breast Cancer Res. Treat. 2011, 129, 69–77. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Kugler, S.; Waddell, N.; Johnstone, C.N.; Marsh, A.; Henderson, S.; Chenevix-Trench, G. DNA methylome of familial breast cancer identifies distinct profiles defined by mutation status. Breast Cancer Res. 2010, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.-J.; Lee, K.-H.; Nam, A.-R.; Cho, J.-Y. Genome-Wide Methylation Profiling in Canine Mammary Tumor Reveals miRNA Candidates Associated with Human Breast Cancer. Cancers 2019, 11, 1466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, Y.; Wang, Y.; Jiang, L.; Li, X.; Gao, H.; Wei, M.; Zhao, L. Integrative Analysis of DNA Methylation and Gene Expression to Determine Specific Diagnostic Biomarkers and Prognostic Biomarkers of Breast Cancer. Front. Cell Dev. Biol. 2020, 8, 529386. [Google Scholar] [CrossRef]
- Mijnes, J.; Tiedemann, J.; Eschenbruch, J.; Gasthaus, J.; Bringezu, S.; Bauerschlag, D.; Dahl, E. SNiPER: A novel hypermethylation biomarker panel for liquid biopsy based early breast cancer detection. Oncotarget 2019, 10, 6494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Danenberg, P.V.; Laird, P.W. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression. Cancer Res. 1999, 59, 2302–2306. [Google Scholar] [PubMed]
- Kanai, Y.; Ushijima, S.; Kondo, Y.; Nakanishi, Y.; Hirohashi, S. DNA methyltransferase expression and DNA methylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int. J. Cancer 2001, 91, 205–212. [Google Scholar] [CrossRef]
- Mizuno, S.-I.; Chijiwa, T.; Okamura, T.; Akashi, K.; Fukumaki, Y.; Niho, Y.; Sasaki, H. Expression of DNA methyltransferases DNMT1,3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001, 97, 1172–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, S.; Patra, A.; Zhao, H.; Dahiya, R. DNA methyltransferase and demethylase in human prostate cancer. Mol. Carcinog. 2002, 33, 163–171. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA Methyltransferases in Cancer: Biology, Paradox, Aberrations, and Targeted Therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef]
- Girault, I.; Tozlu, S.; Lidereau, R.; Bieche, I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin. Cancer Res. 2003, 9, 4415–4422. [Google Scholar]
- Giri, A.K.; Aittokallio, T. DNMT inhibitors increase methylation in the cancer genome. Front. Pharmacol. 2019, 10, 385. [Google Scholar] [CrossRef] [Green Version]
- Lyko, F.; Brown, R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl. Cancer Inst. 2005, 97, 1498–1506. [Google Scholar] [CrossRef]
- Stillman, B. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 2018, 175, 6–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Füllgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Oncogene 2011, 30, 3391–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Lakshmanan, M. Role of novel histone modifications in cancer. Oncotarget 2018, 9, 11414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbert, P.B.; Henikoff, S. Histone variants at a glance. J. Cell Sci. 2021, 134, jcs244749. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global Levels of Histone Modifications Predict Prognosis in Different Cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [Green Version]
- Ruthenburg, A.J.; Li, H.; Patel, D.J.; Allis, C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 2007, 8, 983–994. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.; Zhang, Y. Mechanisms of epigenetic inheritance. Curr. Opin. Cell Biol. 2007, 19, 266–272. [Google Scholar] [CrossRef]
- Tweedie-Cullen, R.Y.; Brunner, A.M.; Grossmann, J.; Mohanna, S.; Sichau, D.; Nanni, P.; Panse, C.; Mansuy, I.M. Identification of Combinatorial Patterns of Post-Translational Modifications on Individual Histones in the Mouse Brain. PLoS ONE 2012, 7, e36980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz Martín, N.; Dave, N.; Millanes Romero, A.; Morey Ramonell, L.; Díaz, V.M.; Lórenz Fonfria, V.; Peiró Sales, S. Lysyl oxidase-like 2 deaminates lysine 4 in histone H3. Mol. Cell 2012, 46, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Byvoet, P.; Shepherd, G.; Hardin, J.; Noland, B. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch. Biochem. Biophys. 1972, 148, 558–567. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, S.; Drozak, J. Protein Histidine Methylation. Curr. Protein Pept. Sci. 2020, 21, 675–689. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Seliga, A.K.; Vertommen, D.; Terreri, M.; Ishikawa, T.; Grabowska, I.; Tiebe, M.; Teleman, A.A.; Jagielski, A.K.; Veiga-Da-Cunha, M.; et al. SETD3 protein is the actin-specific histidine N-methyltransferase. eLife 2018, 7, e37921. [Google Scholar] [CrossRef]
- Upadhyay, A.K.; Cheng, X. Dynamics of Histone Lysine Methylation: Structures of Methyl Writers and Erasers. In Epigenetics and Disease; Springer: Basel, Switzerland, 2011; Volume 67, pp. 107–124. [Google Scholar] [CrossRef]
- Janardhan, A.; Kathera, C.; Darsi, A.; Ali, W.; He, L.; Yang, Y.; Luo, L.; Guo, Z. Prominent role of histone lysine demethylases in cancer epigenetics and therapy. Oncotarget 2018, 9, 34429–34448. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [Green Version]
- Borun, T.W.; Pearson, D.; Paik, W.K. Studies of histone methylation during the HeLa S-3 cell cycle. J. Biol. Chem. 1972, 247, 4288–4298. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Sun, L.; Kokura, K.; Horton, J.; Fukuda, M.; Espejo, A.; Izumi, V.; Koomen, J.M.; Bedford, M.T.; Zhang, X.; et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun. 2011, 2, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res./Rev. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Y.; Zhang, J.; Lu, Y.; Liu, X.; Geng, P.; Huang, B.; Zhang, Y.; Lu, J. The dual function of PRMT1 in modulating epithelial-mesenchymal transition and cellular senescence in breast cancer cells through regulation of ZEB1. Sci. Rep. 2016, 6, 19874. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global Histone Modifications in Breast Cancer Correlate with Tumor Phenotypes, Prognostic Factors, and Patient Outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [Green Version]
- Verdone, L.; Agricola, E.; Caserta, M.; Di Mauro, E. Histone acetylation in gene regulation. Brief Funct Genom. Proteomic 2006, 5, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Adcock, I.; Ito, K. Histone acetylation and deacetylation: Importance in inflammatory lung diseases. Eur. Respir. J. 2005, 25, 552–563. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, T.; Goto, A.; Kurokawa, E.; Hara, H.; Adachi, T. Cross Talk Mechanism among EMT, ROS, and Histone Acetylation in Phorbol Ester-Treated Human Breast Cancer MCF-7 Cells. Oxidative Med. Cell. Longev. 2016, 2016, 1284372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchetti, B.; Shimada, A.K.; Katz, A.; Curigliano, G. The role of histone deacetylase inhibitors in metastatic breast cancer. Breast 2019, 43, 130–134. [Google Scholar] [CrossRef]
- Yang, H.; Salz, T.; Zajac-Kaye, M.; Liao, D.; Huang, S.; Qiu, Y. Overexpression of histone deacetylases in cancer cells is controlled by interplay of transcription factors and epigenetic modulators. FASEB J. 2014, 28, 4265–4279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Z.-W.; Zhou, T.-C.; Tan, X.-J.; Song, X.-L.; Liu, Y.; Shi, X.-Y.; Huang, W.-J.; Du, L.-L.; Tu, B.-J.; Lin, X.-D. High expression of p300 is linked to aggressive features and poor prognosis of Nasopharyngeal Carcinoma. J. Transl. Med. 2012, 10, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Luo, R.-Z.; Chen, J.-W.; Cao, Y.; Lu, J.-B.; He, J.-H.; Wu, Q.-L.; Cai, M.-Y. High expression of transcriptional coactivator p300 correlates with aggressive features and poor prognosis of hepatocellular carcinoma. J. Transl. Med. 2011, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Li, Y.; Luo, R.-Z.; Fu, J.-H.; He, J.-H.; Zhang, L.-J.; Yang, H.-X. High expression of the transcriptional co-activator p300 predicts poor survival in resectable non-small cell lung cancers. Eur. J. Surg. Oncol. (EJSO) 2012, 38, 523–530. [Google Scholar] [CrossRef]
- Isharwal, S.; Miller, M.C.; Marlow, C.; Makarov, D.; Partin, A.W.; Veltri, R.W. p300 (histone acetyltransferase) biomarker predicts prostate cancer biochemical recurrence and correlates with changes in epithelia nuclear size and shape. Prostate 2008, 68, 1097–1104. [Google Scholar] [CrossRef] [Green Version]
- Alsamri, H.; El Hasasna, H.; Baby, B.; Alneyadi, A.; Al Dhaheri, Y.; Ayoub, M.A.; Eid, A.H.; Vijayan, R.; Iratni, R. Carnosol Is a Novel Inhibitor of p300 Acetyltransferase in Breast Cancer. Front. Oncol. 2021, 11, 1650. [Google Scholar] [CrossRef]
- Valor, L.M.; Viosca, J.; Lopez-Atalaya, J.P.; Barco, A. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 2013, 19, 5051–5064. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, A.M.; Wood, M.A.; McDonough, C.B.; Abel, T. Transgenic mice expressing an inhibitory truncated form of p300 exhibit long-term memory deficits. Learn. Mem. 2007, 14, 564–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, A.; Kaur, P.; Lang, J.E. EP300 knockdown reduces cancer stem cell phenotype, tumor growth and metastasis in triple negative breast cancer. BMC Cancer 2020, 20, 1076. [Google Scholar] [CrossRef]
- Zhao, L.; Pang, A.; Li, Y. Function of GCN5 in the TGF-β1-induced epithelial-to-mesenchymal transition in breast cancer. Oncol. Lett. 2018, 16, 3955–3963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, K.; Jeng, W.; Chiang, C.; Chai, C.; Chiou, S.; Huang, M.; Yokoyama, K.K.; Wang, S.; Huang, S.; et al. PCAF-mediated acetylation of ISX recruits BRD 4 to promote epithelial-mesenchymal transition. EMBO Rep. 2020, 21, e48795. [Google Scholar] [CrossRef] [PubMed]
- Majdzadeh, N.; Morrison, B.E.; D’Mello, S.R. Class IIA HDACs in the regulation of neurodegeneration. Front. Biosci. 2008, 13, 1072–1082. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef]
- Kim, E.; Bisson, W.H.; Löhr, C.V.; Williams, D.E.; Ho, E.; Dashwood, R.H.; Rajendran, P. Histone and Non-Histone Targets of Dietary Deacetylase Inhibitors. Curr. Top. Med. Chem. 2016, 16, 714–731. [Google Scholar] [CrossRef]
- Ferrante, F.; Giaimo, B.D.; Bartkuhn, M.; Zimmermann, T.; Close, V.; Mertens, D.; Nist, A.; Stiewe, T.; Meier-Soelch, J.; Kracht, M.; et al. HDAC3 functions as a positive regulator in Notch signal transduction. Nucleic Acids Res. 2020, 48, 3496–3512. [Google Scholar] [CrossRef]
- Leus, N.G.; Zwinderman, M.R.; Dekker, F.J. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-κB-mediated inflammation. Curr. Opin. Chem. Biol. 2016, 33, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 Act Redundantly to Control p63 and p53 Functions in Epidermal Progenitor Cells. Dev. Cell 2010, 19, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA Expression in Invasive Carcinoma of the Breast*. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.-S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone Deacetylase 3 (HDAC3) and Other Class I HDACs Regulate Colon Cell Maturation and p21 Expression and Are Deregulated in Human Colon Cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Byun, D.-S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 Promotes Growth of Colon Cancer Cells via Repression of p21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef] [Green Version]
- Beumer, J.H.; Tawbi, H. Role of histone deacetylases and their inhibitors in cancer biology and treatment. Curr. Clin. Pharmacol. 2010, 5, 196–208. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer-overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [Green Version]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Emerging role of histone deacetylase inhibitors as anti-breast-cancer agents. Drug Discov. Today 2019, 24, 685–702. [Google Scholar] [CrossRef]
- Leslie, P.L.; Chao, Y.L.; Tsai, Y.-H.; Ghosh, S.K.; Porrello, A.; Van Swearingen, A.; Harrison, E.B.; Cooley, B.C.; Parker, J.S.; Carey, L.A.; et al. Histone deacetylase 11 inhibition promotes breast cancer metastasis from lymph nodes. Nat. Commun. 2019, 10, 4192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, C.M. Non-Coding RNAs in Breast Cancer: Intracellular and Intercellular Communication. Non-Coding RNA 2018, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliperti, V.; Skonieczna, J.; Cerase, A. Long Non-Coding RNA (lncRNA) Roles in Cell Biology, Neurodevelopment and Neurological Disorders. Non-Coding RNA 2021, 7, 36. [Google Scholar] [CrossRef]
- Borkiewicz, L.; Kalafut, J.; Dudziak, K.; Przybyszewska-Podstawka, A.; Telejko, I. Decoding LncRNAs. Cancers 2021, 13, 2643. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Y.; Lu, J. The roles of long noncoding RNAs in breast cancer metastasis. Cell Death Dis. 2020, 11, 749. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, L.P.; Lau, N.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Hendrickson, D.G.; Hogan, D.J.; McCullough, H.L.; Myers, J.W.; Herschlag, D.; Ferrell, J.E.; Brown, P.O. Concordant Regulation of Translation and mRNA Abundance for Hundreds of Targets of a Human microRNA. PLoS Biol. 2009, 7, e1000238. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from Repression to Activation: MicroRNAs Can Up-Regulate Translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [Green Version]
- Loh, H.-Y.; Norman, B.P.; Lai, K.-S.; Rahman, N.M.A.N.A.; Alitheen, N.B.M.; Osman, M.A. The Regulatory Role of MicroRNAs in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 4940. [Google Scholar] [CrossRef] [Green Version]
- Kanchan, R.K.; Siddiqui, J.A.; Mahapatra, S.; Batra, S.K.; Nasser, M.W. microRNAs Orchestrate Pathophysiology of Breast Cancer Brain Metastasis: Advances in Therapy. Mol. Cancer 2020, 19, 29. [Google Scholar] [CrossRef] [Green Version]
- Hilton, C.; Neville, M.J.; Karpe, F. MicroRNAs in adipose tissue: Their role in adipogenesis and obesity. Int. J. Obes. 2013, 37, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Quiat, D.; Olson, E.N. MicroRNAs in cardiovascular disease: From pathogenesis to prevention and treatment. J. Clin. Investig. 2013, 123, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef]
- Alajez, N.M.; Lenarduzzi, M.; Ito, E.; Hui, A.B.; Shi, W.; Bruce, J.; Yue, S.; Huang, S.H.; Xu, W.; Waldron, J.; et al. miR-218 Suppresses Nasopharyngeal Cancer Progression through Downregulation of Survivin and the SLIT2-ROBO1 Pathway. Cancer Res. 2011, 71, 2381–2391. [Google Scholar] [CrossRef] [Green Version]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 pathway in cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Alajez, N.; Shi, W.; Wong, D.; Lenarduzzi, M.; Waldron, J.; Weinreb, I.; Liu, F.-F. Lin28b Promotes Head and Neck Cancer Progression via Modulation of the Insulin-Like Growth Factor Survival Pathway. Oncotarget 2012, 3, 1641–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, S.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, I.; Contreras, A.; Cordero, J.; Rubio, K.; Dobersch, S.; Günther, S.; Barreto, G. MiCEE is a ncRNA-protein complex that mediates epigenetic silencing and nucleolar organization. Nat. Genet. 2018, 50, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Wirth, T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Jurmeister, S.; Baumann, M.; Balwierz, A.; Keklikoglou, I.; Ward, A.; Uhlmann, S.; Zhang, J.D.; Wiemann, S.; Sahin, Ö. MicroRNA-200c Represses Migration and Invasion of Breast Cancer Cells by Targeting Actin-Regulatory Proteins FHOD1 and PPM1F. Mol. Cell. Biol. 2012, 32, 633–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A.H. Programmed Cell Death 4 (PDCD4) Is an Important Functional Target of the MicroRNA miR-21 in Breast Cancer Cells. J. Biol. Chem. 2008, 283, 1026–1033. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Si, M.-L.; Wu, H.; Mo, Y.-Y. MicroRNA-21 Targets the Tumor Suppressor Gene Tropomyosin 1 (TPM1). J. Biol. Chem. 2007, 282, 14328–14336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickramasinghe, N.S.; Manavalan, T.T.; Dougherty, S.M.; Riggs, K.A.; Li, Y.; Klinge, C.M. Estradiol downregulates miR-21 expression and increases miR-21 target gene expression in MCF-7 breast cancer cells. Nucleic Acids Res. 2009, 37, 2584–2595. [Google Scholar] [CrossRef] [PubMed]
- Si, M.-L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.-Y. miR-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef]
- Rowland, B.D.; Peeper, D.S. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Cancer 2005, 6, 11–23. [Google Scholar] [CrossRef]
- Png, K.J.; Yoshida, M.; Zhang, X.H.-F.; Shu, W.; Lee, H.; Rimner, A.; Chan, T.A.; Comen, E.; Andrade, V.P.; Kim, S.W.; et al. MicroRNA-335 inhibits tumor reinitiation and is silenced through genetic and epigenetic mechanisms in human breast cancer. Genes Dev. 2011, 25, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Tavazoie, S.F.; Alarcón, C.; Oskarsson, T.; Padua, D.; Wang, Q.; Bos, P.D.; Gerald, W.L.; Massague, J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008, 451, 147–152. [Google Scholar] [CrossRef] [Green Version]
- O’Donovan, P.J.; Livingston, D.M. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C.I.; King, M.-C. Inherited breast and ovarian cancer. Hum. Mol. Genet. 1995, 4 (Suppl. 1), 1811–1817. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, H.-W.; Lu, M.-H.; He, X.-H.; Li, Y.; Gu, H.; Liu, M.-F.; Wang, E.-D. MicroRNA-155 Functions as an OncomiR in Breast Cancer by Targeting the Suppressor of Cytokine Signaling 1 Gene. Cancer Res. 2010, 70, 3119–3127. [Google Scholar] [CrossRef] [Green Version]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yuan, L.; Luo, J.; Gao, J.; Guo, J.; Xie, X. MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. Clin. Exp. Med. 2013, 13, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhu, S.; Mo, Y.-Y. Suppression of cell growth and invasion by miR-205 in breast cancer. Cell Res. 2009, 19, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Casalini, P.; Piovan, C.; Di Leva, G.; Merlo, A.; Triulzi, T.; Ménard, S.; Croce, C.M.; Tagliabue, E. microRNA-205 Regulates HER3 in Human Breast Cancer. Cancer Res. 2009, 69, 2195–2200. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Issah, M.A.; Wu, D.; Zhang, F.; Zheng, W.; Liu, Y.; Fu, H.; Zhou, H.; Chen, R.; Shen, J. Epigenetic modifications in acute myeloid leukemia: The emerging role of circular RNAs (Review). Int. J. Oncol. 2021, 59, 107. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, X.; Xu, J.; Li, Q.; Hao, L.; Zeng, Z.; Xiao, M.; Song, J.; Liu, F.; Fang, C.; et al. Identification and characterization of N6-methyladenosine modification of circRNAs in glioblastoma. J. Cell. Mol. Med. 2021, 25, 7204–7217. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef]
- Liu, T.; Wei, Q.; Jin, J.; Luo, Q.; Liu, Y.; Yang, Y.; Cheng, C.; Li, L.; Pi, J.; Si, Y.; et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020, 48, 3816–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Jaffrey, S.R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014, 15, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Klinge, C.M.; Piell, K.M.; Tooley, C.S.; Rouchka, E.C. HNRNPA2/B1 is upregulated in endocrine-resistant LCC9 breast cancer cells and alters the miRNA transcriptome when overexpressed in MCF-7 cells. Sci. Rep. 2019, 9, 9430. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m6A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [Green Version]
- Yi, D.; Wang, R.; Shi, X.; Xu, L.; Yilihamu, Y.E.; Sang, J. METTL14 promotes the migration and invasion of breast cancer cells by modulating N6-methyladenosine and hsa-miR-146a-5p expression. Oncol. Rep. 2020, 43, 1375–1386. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Kuo, H.-C. The emerging roles and functions of circular RNAs and their generation. J. Biomed. Sci. 2019, 26, 29. [Google Scholar] [CrossRef]
- Nigro, J.M.; Cho, K.R.; Fearon, E.R.; Kern, S.E.; Ruppert, J.M.; Oliner, J.D.; Vogelstein, B. Scrambled exons. Cell 1991, 64, 607–613. [Google Scholar] [CrossRef]
- Capel, B.; Swain, A.; Nicolis, S.; Hacker, A.; Walter, M.; Koopman, P.; Goodfellow, P.; Lovell-Badge, R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993, 73, 1019–1030. [Google Scholar] [CrossRef]
- Cocquerelle, C.; Daubersies, P.; Majerus, M.A.; Kerckaert, J.P.; Bailleul, B. Splicing with inverted order of exons occurs proximal to large introns. EMBO J. 1992, 11, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Xie, X.; Li, M.; Shi, J.; Zhou, J.J.; Knox, K.S.; Wang, T.; Chen, Q.; Gu, W. Rat BodyMap transcriptomes reveal unique circular RNA features across tissue types and developmental stages. RNA 2018, 24, 1443–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.-J.; Yin, J.-W.; Wu, J.-H.; Gu, J.; Yuan, C.-Y.; Miao, H.-J.; Yu, Z.-B. Circular RNAs are abundant and dynamically expressed during the embryonic lung development of C57BL/6 mice. Heliyon 2020, 6, e03437. [Google Scholar] [CrossRef]
- Misir, S.; Wu, N.; Yang, B.B. Specific expression and functions of circular RNAs. Cell Death Differ. 2022, 29, 481–491. [Google Scholar] [CrossRef]
- Chen, X.; Han, P.; Zhou, T.; Guo, X.; Song, X.; Li, Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016, 6, 34985. [Google Scholar] [CrossRef]
- Zhou, W.Y.; Cai, Z.R.; Liu, J.; Wang, D.S.; Ju, H.Q.; Xu, R.H. Circular RNA: Metabolism, functions and interactions with proteins. Mol. Cancer 2020, 19, 172. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Wang, Z. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef] [Green Version]
- Di Timoteo, G.; Dattilo, D.; Centrón-Broco, A.; Colantoni, A.; Guarnacci, M.; Rossi, F.; Incarnato, D.; Oliviero, S.; Fatica, A.; Morlando, M.; et al. Modulation of circRNA Metabolism by m6A Modification. Cell Rep. 2020, 31, 107641. [Google Scholar] [CrossRef]
- Wang, L.; Long, H.; Zheng, Q.; Bo, X.; Xiao, X.; Li, B. Circular RNA circRHOT1 promotes hepatocellular carcinoma progression by initiation of NR2F6 expression. Mol. Cancer 2019, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Ma, J.; Sun, T.; Zhou, Q.; Wang, W.; Wang, G.; Wu, P.; Wang, H.; Jiang, L.; et al. Exosomal circRNAs: Biogenesis, effect and application in human diseases. Mol. Cancer 2019, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.R.; Munk, R.; Kundu, G.; De, S.; Abdelmohsen, K.; Gorospe, M. Methods for analysis of circular RNAs. Wiley Interdiscip. Rev. RNA 2020, 11, e1566. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.O.; Wei, W.; Huang, X.; Xie, X.; Kong, Y.; Dai, D.; Xie, X. circEPSTI1 as a prognostic marker and mediator of triple-negative breast cancer progression. Theranostics 2018, 8, 4003. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, Y.; Liang, G.; Ling, Y.; Tan, W.; Tan, L.; Andrews, R.; Zhong, W.; Zhang, X.; Song, E.; et al. Circular RNA hsa_circ_001783 regulates breast cancer progression via sponging miR-200c-3p. Cell Death Dis. 2019, 10, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsburg, O.; Yip, C.; Brooks, A.; Cabanes, A.; Caleffi, M.; Yataco, J.A.D.; Gyawali, B.; McCormack, V.; de Anderson, M.M.; Mehrotra, R.; et al. Breast cancer early detection: A phased approach to implementation. Cancer 2020, 126, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Jedy-Agba, E.; McCormack, V.; Adebamowo, C.; Dos-Santos-Silva, I. Stage at diagnosis of breast cancer in sub-Saharan Africa: A systematic review and meta-analysis. Lancet Glob. Health 2016, 4, e923–e935. [Google Scholar] [CrossRef] [Green Version]
- Justo, N.; Wilking, N.; Jönsson, B.; Luciani, S.; Cazap, E. A Review of Breast Cancer Care and Outcomes in Latin America. Oncology 2013, 18, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Unger-Saldaña, K.; Miranda, A.; Zarco-Espinosa, G.; Mainero-Ratchelous, F.; Bargalló-Rocha, E.; Lázaro-León, J.M. Health system delay and its effect on clinical stage of breast cancer: Multicenter study. Cancer 2015, 121, 2198–2206. [Google Scholar] [CrossRef]
- Caplan, L. Delay in breast cancer: Implications for stage at diagnosis and survival. Front. Public Health 2014, 2, 87. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.J.S. Early diagnosis of breast cancer. Sensors 2017, 17, 1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Guan, X.; Fan, Z.; Ching, L.-M.; Li, Y.; Wang, X.; Cao, W.-M.; Liu, D.-X. Non-Invasive Biomarkers for Early Detection of Breast Cancer. Cancers 2020, 12, 2767. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Shi, G.; He, Q.; Zhu, P. Screening and predicted value of potential biomarkers for breast cancer using bioinformatics analysis. Sci. Rep. 2021, 11, 20799. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Fasching, P.A.; Beckmann, M.W.; Kölbl, H. Biomarkers in breast cancer–an update. Geburtshilfe Frauenheilkd. 2012, 72, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Louie, A.D.; Huntington, K.; Carlsen, L.; Zhou, L.; El-Deiry, W.S. Integrating Molecular Biomarker Inputs Into Development and Use of Clinical Cancer Therapeutics. Front. Pharmacol. 2021, 12, 2850. [Google Scholar] [CrossRef]
- Alba-Bernal, A.; Lavado-Valenzuela, R.; Domínguez-Recio, M.E.; Jiménez-Rodriguez, B.; Queipo-Ortuño, M.I.; Alba, E.; Comino-Méndez, I. Challenges and achievements of liquid biopsy technologies employed in early breast cancer. eBioMedicine 2020, 62, 103100. [Google Scholar] [CrossRef]
- Kim, J.-H.; Shin, M.-H.; Kweon, S.-S.; Park, M.H.; Yoon, J.H.; Lee, J.S.; Choi, C.; Fackler, M.J.; Sukumar, S. Evaluation of promoter hypermethylation detection in serum as a diagnostic tool for breast carcinoma in Korean women. Gynecol. Oncol. 2010, 118, 176–181. [Google Scholar] [CrossRef]
- Salta, S.; Nunes, S.P.; Fontes-Sousa, M.; Lopes, P.; Freitas, M.; Caldas, M.; Antunes, L.; Castro, F.; Antunes, P.; de Sousa, S.P.; et al. A DNA Methylation-Based Test for Breast Cancer Detection in Circulating Cell-Free DNA. J. Clin. Med. 2018, 7, 420. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, B.P.; Apolónio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer 2019, 19, 219. [Google Scholar] [CrossRef] [Green Version]
- Micalizzi, D.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.; Huang, X.; Woodcock, M.; Du, M.; Dittmar, R.; Wang, Y.; Tsai, S.; Kohli, M.; Boardman, L.; Patel, T.; et al. Plasma extracellular RNA profiles in healthy and cancer patients. Sci. Rep. 2016, 6, srep19413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Fan, J.; Hsu, Y.-M.S.; Lyon, C.J.; Ning, B.; Hu, T.Y. Extracellular vesicles as cancer liquid biopsies: From discovery, validation, to clinical application. Lab Chip 2019, 19, 1114–1140. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Li, Y.; Wang, M.; Gu, J.; Xu, W.; Cai, H.; Fang, X.; Zhang, X. Exosomes as a new frontier of cancer liquid biopsy. Mol. Cancer 2022, 21, 56. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- García-Pardo, M.; Makarem, M.; Li, J.J.N.; Kelly, D.; Leighl, N.B. Integrating circulating-free DNA (cfDNA) analysis into clinical practice: Opportunities and challenges. Br. J. Cancer 2022, 127, 592–602. [Google Scholar] [CrossRef]
- Zhao, X.; Dai, F.; Mei, L.; Huang, D.; Shen, X.; Zhang, H.; She, X.; Ma, Z. The Potential Use of Dynamics Changes of ctDNA and cfDNA in the Perioperative Period to Predict the Recurrence Risk in Early NSCLC. Front. Oncol. 2021, 11, 2499. [Google Scholar] [CrossRef]
- Czeiger, D.; Shaked, G.; Sebbag, G.; Vakhrushev, A.; Flomboym, A.; Lior, Y.; Belochitski, O.; Ariad, S.; Douvdevani, A. Elevated Cell-Free DNA Measured by a Simple Assay Is Associated With Increased Rate of Colorectal Cancer Relapse. Am. J. Clin. Pathol. 2016, 145, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.F.; Hills, A.; Page, K.; Hastings, R.K.; Toghill, B.; Goddard, K.S.; Ion, C.; Ogle, O.; Boydell, A.R.; Gleason, K.; et al. Plasma cell-free DNA (cfDNA) as a predictive and prognostic marker in patients with metastatic breast cancer. Breast Cancer Res. 2019, 21, 149. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E.; et al. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef]
- Peled, M.; Agassi, R.; Czeiger, D.; Ariad, S.; Riff, R.; Rosenthal, M.; Lazarev, I.; Novack, V.; Yarza, S.; Mizrakli, Y.; et al. Cell-free DNA concentration in patients with clinical or mammographic suspicion of breast cancer. Sci. Rep. 2020, 10, 14601. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Hu, Y.; Li, Q.; Chen, X.; Huang, J.; Pellegrini, M.; Zhou, X.J.; Rettig, M.; Fan, G. The size of cell-free mitochondrial DNA in blood is inversely correlated with tumor burden in cancer patients. Precis. Clin. Med. 2019, 2, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, G.C.; Petrone, A.B.; Tennant, C.S.; Lucke-Wold, N.; Kabbani, Y.; Tarabishy, A.R.; Chantler, P.D.; Barr, T.L. Circulating extracellular DNA levels are acutely elevated in ischaemic stroke and associated with innate immune system activation. Brain Inj. 2017, 31, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.; Burchell, R.; Worth, A.; Burton, S.; Gedye, K.; Clark, K.; Crosse, K.; Jack, M.; Odom, T.; De Grey, S.; et al. Kinetics of Plasma Cell-Free DNA and Creatine Kinase in a Canine Model of Tissue Injury. J. Vet. Intern. Med. 2018, 32, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C.C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Marigorta, U.M.; Rodríguez, J.A.; Gibson, G.; Navarro, A. Replicability and Prediction: Lessons and Challenges from GWAS. Trends Genet. 2018, 34, 504–517. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-X.; Chen, H.; Rong, Y.; Jiang, F.; Chen, J.-B.; Duan, Y.-Y.; Zhu, D.-L.; Yang, T.-L.; Dai, Z.; Dong, S.-S.; et al. An integrative multi-omics network-based approach identifies key regulators for breast cancer. Comput. Struct. Biotechnol. J. 2020, 18, 2826–2835. [Google Scholar] [CrossRef]
- Menyhárt, O.; Győrffy, B. Multi-omics approaches in cancer research with applications in tumor subtyping, prognosis, and diagnosis. Comput. Struct. Biotechnol. J. 2021, 19, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wu, Y.; Cheng, W.; Wu, Y.; Wang, L.; Zhuang, L. Identification of novel prognostic biomarkers by integrating multi-omics data in gastric cancer. BMC Cancer 2021, 21, 460. [Google Scholar] [CrossRef] [PubMed]
- Lech, G.; Slotwinski, R.; Słodkowski, M.; Krasnodębski, I.W. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J. Gastroenterol. 2016, 22, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-T.; Chuang, Y.-M.; Chan, M.W.Y. Combinatorial Epigenetic and Immunotherapy in Breast Cancer Management: A Literature Review. Epigenomes 2020, 4, 27. [Google Scholar] [CrossRef]
- Abdel-Hafiz, H.A.; Horwitz, K.B. Role of epigenetic modifications in luminal breast cancer. Epigenomics 2015, 7, 847–862. [Google Scholar] [CrossRef]
- Kamińska, K.; Nalejska, E.; Kubiak, M.; Wojtysiak, J.; Żołna, Ł.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Epigenetic Biomarkers in Oncology. Mol. Diagn. Ther. 2019, 23, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenetics 2021, 13, 166. [Google Scholar] [CrossRef]
- Khandelwal, M.; Anand, V.; Appunni, S.; Seth, A.; Singh, P.; Mathur, S.; Sharma, A. Decitabine augments cytotoxicity of cisplatin and doxorubicin to bladder cancer cells by activating hippo pathway through RASSF1A. Mol. Cell. Biochem. 2018, 446, 105–114. [Google Scholar] [CrossRef]
- Pinto, A.; Maio, M.; Attadia, V.; Zappacosta, S.; Cimino, R. Modulation of Hla-Dr Antigens Expression in Human Myeloid Leukaemia Cells by Cytarabine and 5-Aza-2’-Deoxycytidine. Lancet 1984, 324, 867–868. [Google Scholar] [CrossRef]
- Lapidus, R.G.; Ferguson, A.T.; Ottaviano, Y.L.; Parl, F.F.; Smith, H.S.; Weitzman, S.A.; Baylin, S.B.; Issa, J.P.; Davidson, N.E. Methylation of estrogen and progesterone receptor gene 5’ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin. Cancer Res. 1996, 2, 805–810. [Google Scholar]
- Zhou, Q.; Atadja, P.; Davidson, N.E. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol. Ther. 2007, 6, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahn, M.L.; Cruickshank, B.M.; Jackson, A.J.; Dean, C.; Holloway, R.W.; Hall, S.R.; Coyle, K.M.; Maillet, H.; Waisman, D.M.; Goralski, K.B.; et al. Decitabine Response in Breast Cancer Requires Efficient Drug Processing and Is Not Limited by Multidrug Resistance. Mol. Cancer Ther. 2020, 19, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenetics 2017, 9, 59. [Google Scholar] [CrossRef]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrump, D.S. Cytotoxicity Mediated by Histone Deacetylase Inhibitors in Cancer Cells: Mechanisms and Potential Clinical Implications. Clin. Cancer Res. 2009, 15, 3947–3957. [Google Scholar] [CrossRef] [Green Version]
- Gjaltema, R.A.; Rots, M.G. Advances of epigenetic editing. Curr. Opin. Chem. Biol. 2020, 57, 75–81. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Abuhamad, A.Y.; Zamberi, N.N.M.; Sheen, L.; Naes, S.M.; Yusuf, S.N.H.M.; Tajudin, A.A.; Mohtar, M.A.; Hamzah, A.S.A.; Syafruddin, S.E. Reverting TP53 Mutation in Breast Cancer Cells: Prime Editing Workflow and Technical Considerations. Cells 2022, 11, 1612. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Scholefield, J.; Harrison, P.T. Prime editing–an update on the field. Gene Ther. 2021, 28, 396–401. [Google Scholar] [CrossRef]
- Jiang, Y.-Y.; Chai, Y.-P.; Lu, M.-H.; Han, X.-L.; Lin, Q.; Zhang, Y.; Zhang, Q.; Zhou, Y.; Wang, X.-C.; Gao, C.; et al. Prime editing efficiently generates W542L and S621I double mutations in two ALS genes in maize. Genome Biol. 2020, 21, 257. [Google Scholar] [CrossRef]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.M.; van Boxtel, R.; van der Doef, H.P.J.; van der Laan, L.J.W.; Verstegen, M.M.A.; van Hasselt, P.M.; et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020, 11, 5352. [Google Scholar] [CrossRef]
- Gao, P.; Lyu, Q.; Ghanam, A.R.; Lazzarotto, C.R.; Newby, G.A.; Zhang, W.; Choi, M.; Slivano, O.J.; Holden, K.; Walker, J.A.; et al. Prime editing in mice reveals the essentiality of a single base in driving tissue-specific gene expression. Genome Biol. 2021, 22, 83. [Google Scholar] [CrossRef]
- Kim, H.K.; Yu, G.; Park, J.; Min, S.; Lee, S.; Yoon, S.; Kim, H.H. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 2021, 39, 198–206. [Google Scholar] [CrossRef]
- Sürün, D.; Schneider, A.; Mircetic, J.; Neumann, K.; Lansing, F.; Paszkowski-Rogacz, M.; Hänchen, V.; Lee-Kirsch, M.A.; Buchholz, F. Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors. Genes 2020, 11, 511. [Google Scholar] [CrossRef]
- Rousseau, J.; Mbakam, C.H.; Guyon, A.; Tremblay, G.; Begin, F.G.; Tremblay, J.P. Specific mutations in genes responsible for Alzheimer and for Duchenne muscular dystrophy introduced by base editing and PRIME editing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Geurts, M.H.; de Poel, E.; Pleguezuelos-Manzano, C.; Oka, R.; Carrillo, L.; Andersson-Rolf, A.; Boretto, M.; Brunsveld, J.E.; van Boxtel, R.; Beekman, J.M.; et al. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Sci. Alliance 2021, 4, e202000940. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Gong, Y.; Jin, M.; Hu, X.; Di, G.; Shao, Z. In vivo multidimensional CRISPR screens identify LGALS2 as an immunotherapy target in triple-negative breast cancer. Sci. Adv. 2022, 26, 8247. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Yan, G.; Wang, N.; Daliah, G.; Edick, A.M.; Poulet, S.; Boudreault, J.; Ali, S.; Burgos, S.A.; Lebrun, J.-J. In vivo genome-wide CRISPR screen reveals breast cancer vulnerabilities and synergistic mTOR/Hippo targeted combination therapy. Nat. Commun. 2021, 12, 3055. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Menz, J.; Modrzejewski, D.; Hartung, F.; Wilhelm, R.; Sprink, T. Genome Edited Crops Touch the Market: A View on the Global Development and Regulatory Environment. Front. Plant Sci. 2020, 11, 586027. [Google Scholar] [CrossRef]
- Wang, H.; Hu, Y.-C.; Markoulaki, S.; Welstead, G.G.; Cheng, A.W.; Shivalila, C.S.; Pyntikova, T.; Dadon, D.B.; Voytas, D.F.; Bogdanove, A.J.; et al. TALEN-mediated editing of the mouse Y chromosome. Nat. Biotechnol. 2013, 31, 530–532. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Zhong, Y.; Li, X.; Li, Y.; Li, X.; Cao, J.; Fan, Z.; Fan, H.; Yuan, L.; Xu, B.; et al. Generation of TALEN-mediated FH knockout rat model. Oncotarget 2016, 7, 61656–61669. [Google Scholar] [CrossRef] [Green Version]
- Carlson, D.F.; Tan, W.; Lillico, S.G.; Stverakova, D.; Proudfoot, C.; Christian, M.; Voytas, D.F.; Long, C.R.; Whitelaw, C.B.A.; Fahrenkrug, S.C. Efficient TALEN-mediated gene knockout in livestock. Proc. Natl. Acad. Sci. USA 2012, 109, 17382–17387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarvari, P.; Rasouli, S.J.; Allanki, S.; Stone, O.A.; Sokol, A.M.; Graumann, J.; Stainier, D.Y. The E3 ubiquitin-protein ligase Rbx1 regulates cardiac wall morphogenesis in zebrafish. Dev. Biol. 2021, 480, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Boch, J. TALE and TALEN genome editing technologies. Gene Genome Ed. 2021, 2, 100007. [Google Scholar] [CrossRef]
- Osborn, M.J.; Starker, C.; McElroy, A.N.; Webber, B.R.; Riddle, M.J.; Xia, L.; DeFeo, A.P.; Gabriel, R.; Schmidt, M.; Von Kalle, C.; et al. TALEN-based Gene Correction for Epidermolysis Bullosa. Mol. Ther. 2013, 21, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Berdien, B.; Abramowski-Mock, U.; Atanackovic, D.; Fehse, B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014, 21, 539–548. [Google Scholar] [CrossRef]
- Mock, U.; Machowicz, R.; Hauber, I.; Horn, S.; Abramowski, P.; Berdien, B.; Fehse, B. mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res. 2015, 43, 5560–5571. [Google Scholar] [CrossRef]
- Cai, Y.; Bak, R.O.; Mikkelsen, J.G. Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife 2014, 3, e01911. [Google Scholar] [CrossRef]
- Holkers, M.; Maggio, I.; Liu, J.; Janssen, J.M.; Miselli, F.; Mussolino, C.; Recchia, A.; Cathomen, T.; Gonçalves, M.A.F.V. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013, 41, e63. [Google Scholar] [CrossRef]
- Chamorro, C.; Mencía, A.; Almarza, D.; Duarte, B.; Büning, H.; Sallach, J.; Hausser, I.; Del Río, M.; Larcher, F.; Murillas, R. Gene Editing for the Efficient Correction of a Recurrent COL7A1 Mutation in Recessive Dystrophic Epidermolysis Bullosa Keratinocytes. Mol. Ther. Nucleic Acids 2016, 5, e307. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Jin, Y.; Bian, T.; Wu, D.; Yang, L.; Terada, N.; Wu, W.; Jin, S. Bacterial Delivery of TALEN Proteins for Human Genome Editing. PLoS ONE 2014, 9, e91547. [Google Scholar] [CrossRef]
- Jia, J.; Bai, F.; Jin, Y.; Santostefano, K.E.; Ha, U.-H.; Wu, D.; Wu, W.; Terada, N.; Jin, S. Efficient Gene Editing in Pluripotent Stem Cells by Bacterial Injection of Transcription Activator-Like Effector Nuclease Proteins. STEM CELLS Transl. Med. 2015, 4, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.U.; Chung, J.Y.; Kim, Y.-H. Current and future delivery systems for engineered nucleases: ZFN, TALEN and RGEN. J. Control. Release 2015, 205, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayoral-Peña, K.; Peña, O.I.G.; Clark, A.M.O.; Flores-Vallejo, R.D.C.; Oza, G.; Sharma, A.; De Donato, M. Biorecognition Engineering Technologies for Cancer Diagnosis: A Systematic Literature Review of Non-Conventional and Plausible Sensor Development Methods. Cancers 2022, 14, 1867. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Varty, K.; O’Brien, C.; Ignaszak, A. Breast Cancer Aptamers: Current Sensing Targets, Available Aptamers, and Their Evaluation for Clinical Use in Diagnostics. Cancers 2021, 13, 3984. [Google Scholar] [CrossRef]
- Subramanian, N.; Kanwar, J.R.; Kanwar, R.K.; Sreemanthula, J.; Biswas, J.; Khetan, V.; Krishnakumar, S. EpCAM aptamer-siRNA chimera targets and regress epithelial cancer. PLoS ONE 2015, 10, e0132407. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Leonard, M.; Shu, Y.; Yang, Y.; Shu, D.; Guo, P.; Zhang, X. Overcoming Tamoxifen Resistance of Human Breast Cancer by Targeted Gene Silencing Using Multifunctional pRNA Nanoparticles. ACS Nano 2017, 11, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Xu, T.; Altai, M.; Orougeni, M.; Zhang, J.; Vorobyeva, A.; Vorontsova, O.; Vtorushin, S.V.; Tolmachev, V.; Gräslund, T.; et al. The Influence of Domain Permutations of an Albumin-Binding Domain-Fused HER2-Targeting Affibody-Based Drug Conjugate on Tumor Cell Proliferation and Therapy Efficacy. Pharmaceutics 2021, 13, 1974. [Google Scholar] [CrossRef]
- Xia, X.; Yang, X.; Huang, W.; Xia, X.; Yan, D. Self-Assembled Nanomicelles of Affibody-Drug Conjugate with Excellent Therapeutic Property to Cure Ovary and Breast Cancers. Nano-Micro Lett. 2022, 14, 33. [Google Scholar] [CrossRef]
- Yamaguchi, H.; On, J.; Morita, T.; Suzuki, T.; Okada, Y.; Ono, J.; Evdokiou, A. Combination of Near-Infrared Photoimmunotherapy Using Trastuzumab and Small Protein Mimetic for HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2021, 22, 12213. [Google Scholar] [CrossRef] [PubMed]
- Maruoka, Y.; Wakiyama, H.; Choyke, P.L.; Kobayashi, H. Near infrared photoimmunotherapy for cancers: A translational perspective. eBioMedicine 2021, 70, 103501. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Author response: Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef] [PubMed]
- Nagalingam, A.; Tighiouart, M.; Ryden, L.; Joseph, L.; Landberg, G.; Saxena, N.K.; Sharma, D. Med1 plays a critical role in the development of tamoxifen resistance. Carcinogenesis 2012, 33, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Lin, Q. Estrogen Receptor Bio-Activities Determine Clinical Endocrine Treatment Options in Estrogen Receptor-Positive Breast Cancer. Technol. Cancer Res. Treat. 2022, 21, 15330338221090351. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.P.; Roberts, C.W. Partitioning of Chemotherapeutics into Nuclear Condensates—Opening the Door to New Approaches for Drug Development. Mol. Cell 2020, 79, 544–545. [Google Scholar] [CrossRef]
- Klein, I.A.; Boija, A.; Afeyan, L.K.; Hawken, S.W.; Fan, M.; Dall’Agnese, A.; Young, R.A. Partitioning of cancer therapeutics in nuclear condensates. Science 2020, 368, 1386–1392. [Google Scholar] [CrossRef]
- Spegg, V.; Altmeyer, M. Biomolecular condensates at sites of DNA damage: More than just a phase. DNA Repair 2021, 106, 103179. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Igelmann, S.; Lessard, F.; Ferbeyre, G. Liquid–Liquid Phase Separation in Cancer Signaling, Metabolism and Anticancer Therapy. Cancers 2022, 14, 1830. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Lu, W.; Mezhuev, Y.; Lan, M.; Li, L.; Liu, F.; Cai, T.; Wu, X.; Cai, Y. A review of nanoparticle drug delivery systems responsive to endogenous breast cancer microenvironment. Eur. J. Pharm. Biopharm. 2021, 166, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Gavas, S.; Quazi, S.; Karpiński, T.M. Nanoparticles for Cancer Therapy: Current Progress and Challenges. Nanoscale Res. Lett. 2021, 16, 173. [Google Scholar] [CrossRef]
- Liu, J.-P.; Wang, T.-T.; Wang, D.-G.; Dong, A.-J.; Li, Y.-P.; Yu, H.-J. Smart nanoparticles improve therapy for drug-resistant tumors by overcoming pathophysiological barriers. Acta Pharmacol. Sin. 2017, 38, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zang, X.; Zhao, X.; Hu, H.; Qiao, M.; Deng, Y.; Chen, D. Nanoparticles for tumor immunotherapy. Eur. J. Pharm. Biopharm. 2017, 115, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Zhang, L.; Chan, J.M.; Gu, F.X.; Rhee, J.-W.; Wang, A.Z.; Radovic-Moreno, A.F.; Alexis, F.; Langer, R.; Farokhzad, O.C. Self-Assembled Lipid−Polymer Hybrid Nanoparticles: A Robust Drug Delivery Platform. ACS Nano 2008, 2, 1696–1702. [Google Scholar] [CrossRef] [Green Version]
- Omidi, Y.; Barar, J. Targeting tumor microenvironment: Crossing tumor interstitial fluid by multifunctional nanomedicines. BioImpacts 2014, 4, 55–67. [Google Scholar] [CrossRef]
- Muhamad, N.; Plengsuriyakarn, T.; Na-Bangchang, K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: A systematic review. Int. J. Nanomed. 2018, 13, 3921–3935. [Google Scholar] [CrossRef] [Green Version]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, R.N.; VasanthaKumar, S.; Bende, G.; Snehalatha, M. Nanoparticulate drug delivery systems for cancer chemotherapy. Mol. Membr. Biol. 2010, 27, 215–231. [Google Scholar] [CrossRef]
- Shi, J.; Xiao, Z.; Kamaly, N.; Farokhzad, O.C. Self-Assembled Targeted Nanoparticles: Evolution of Technologies and Bench to Bedside Translation. Accounts Chem. Res. 2011, 44, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.H. Drug targeting and tumor heterogeneity. J. Control. Release Off. J. Control. Release Soc. 2009, 133, 2. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198. [Google Scholar] [CrossRef] [Green Version]
- Nag, O.K.; Delehanty, J.B. Active Cellular and Subcellular Targeting of Nanoparticles for Drug Delivery. Pharmaceutics 2019, 11, 543. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46 Pt 1, 6387–6392. [Google Scholar]
- Swartz, M.A.; Fleury, M.E. Interstitial Flow and Its Effects in Soft Tissues. Annu. Rev. Biomed. Eng. 2007, 9, 229–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padera, T.P.; Stoll, B.R.; Tooredman, J.B.; Capen, D.; Di Tomaso, E.; Jain, R.K. Cancer cells compress intratumour vessels. Nature 2004, 427, 695. [Google Scholar] [CrossRef] [PubMed]
- Decuzzi, P.; Pasqualini, R.; Arap, W.; Ferrari, M. Intravascular Delivery of Particulate Systems: Does Geometry Really Matter? Pharm. Res. 2009, 26, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturoli, D.; Rippe, B. Ficoll and dextran vs. globular proteins as probes for testing glomerular permselectivity: Effects of molecular size, shape, charge, and deformability. Am. J. Physiol. -Ren. Physiol. 2005, 288, F605–F613. [Google Scholar] [CrossRef]
- Yang, Q.; Jones, S.W.; Parker, C.L.; Zamboni, W.C.; Bear, J.E.; Lai, S.K. Evading Immune Cell Uptake and Clearance Requires PEG Grafting at Densities Substantially Exceeding the Minimum for Brush Conformation. Mol. Pharm. 2014, 11, 1250–1258. [Google Scholar] [CrossRef]
- Perrault, S.D.; Walkey, C.; Jennings, T.; Fischer, H.C.; Chan, W.C.W. Mediating Tumor Targeting Efficiency of Nanoparticles Through Design. Nano Lett. 2009, 9, 1909–1915. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Zhang, M.; Kumar, S.; Vogus, D.R.; Menegatti, S.; Helgeson, M.E.; Mitragotri, S. Elasticity of Nanoparticles Influences Their Blood Circulation, Phagocytosis, Endocytosis, and Targeting. ACS Nano 2015, 9, 3169–3177. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarvari, P.; Sarvari, P.; Ramírez-Díaz, I.; Mahjoubi, F.; Rubio, K. Advances of Epigenetic Biomarkers and Epigenome Editing for Early Diagnosis in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 9521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179521
Sarvari P, Sarvari P, Ramírez-Díaz I, Mahjoubi F, Rubio K. Advances of Epigenetic Biomarkers and Epigenome Editing for Early Diagnosis in Breast Cancer. International Journal of Molecular Sciences. 2022; 23(17):9521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179521
Chicago/Turabian StyleSarvari, Pourya, Pouya Sarvari, Ivonne Ramírez-Díaz, Frouzandeh Mahjoubi, and Karla Rubio. 2022. "Advances of Epigenetic Biomarkers and Epigenome Editing for Early Diagnosis in Breast Cancer" International Journal of Molecular Sciences 23, no. 17: 9521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179521