


HCA (2-Hydroxy-Docosahexaenoic Acid) Induces Apoptosis and Endoplasmic Reticulum Stress in Pancreatic Cancer Cells
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
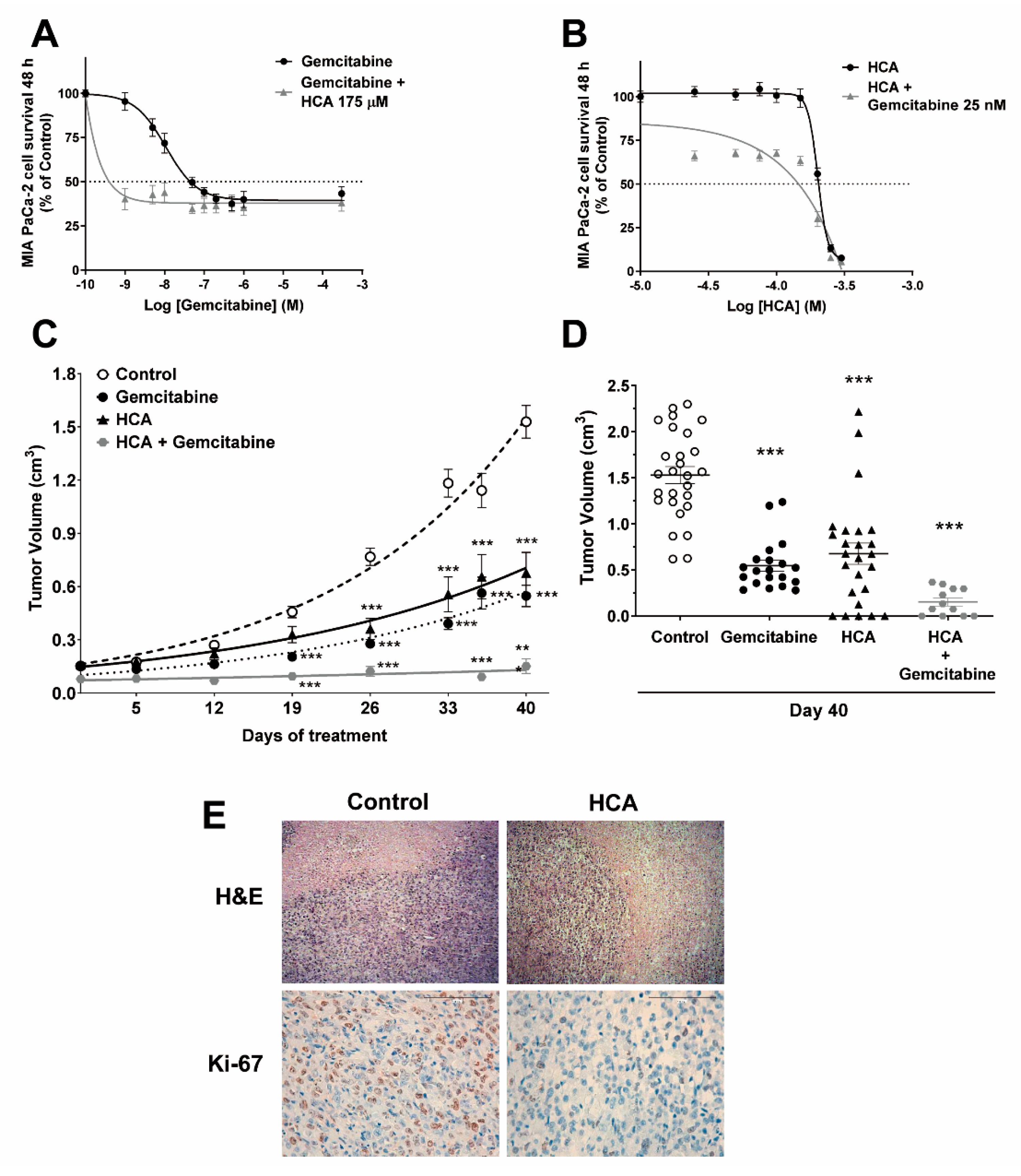
2.1. Efficacy of HCA against Pancreatic Cancer
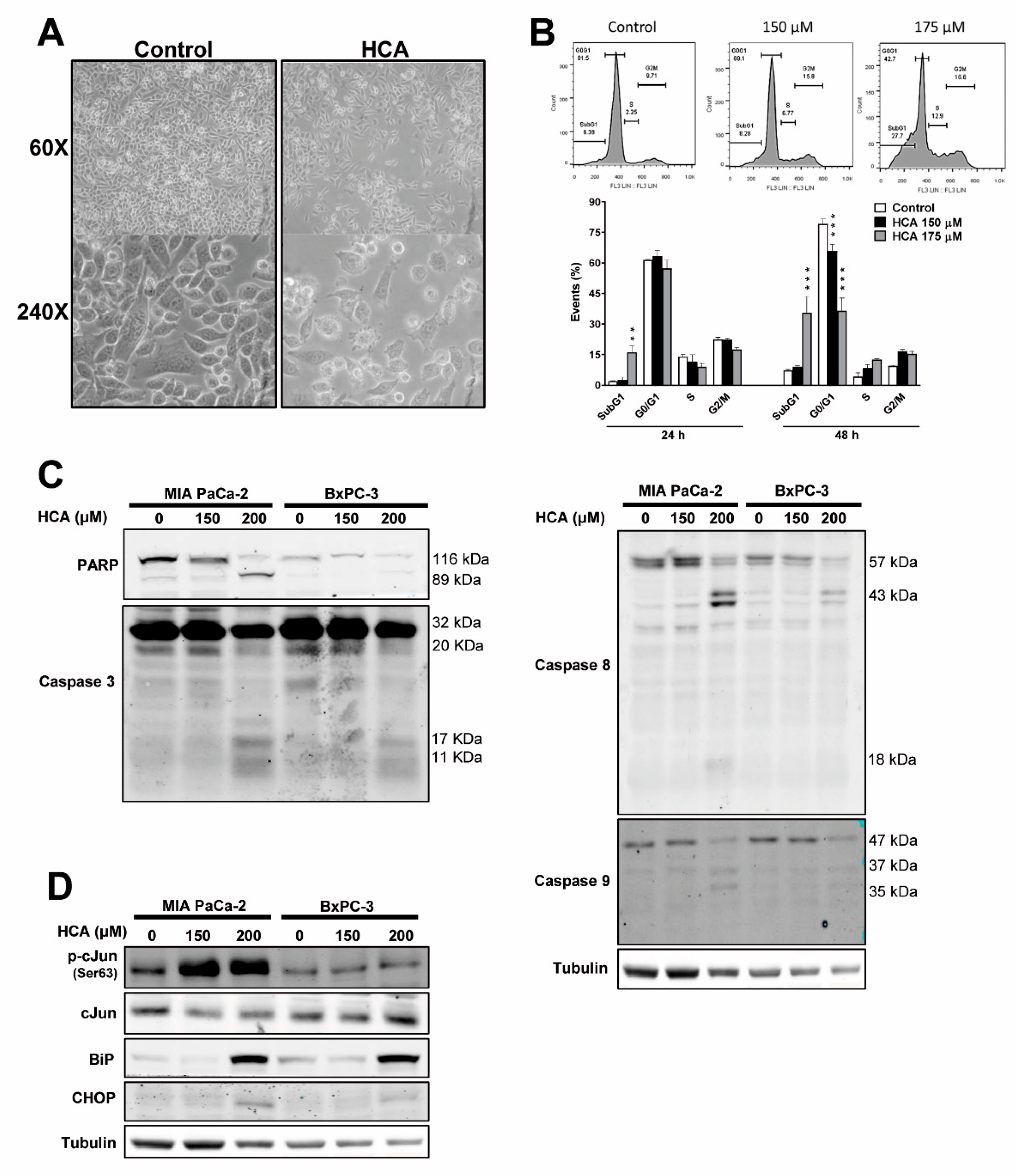
2.2. HCA Induces Intrinsic and Extrinsic Apoptosis, and ER Stress/UPR Signaling in Pancreatic Cancer Cells
2.3. HCA Enhances Caspase-3/7 Activity and Mainly Induces Apoptosis of Pancreatic Cancer Cells through the Extrinsic Pathway
2.4. HCA Causes an Increase in Reactive Oxygen Species
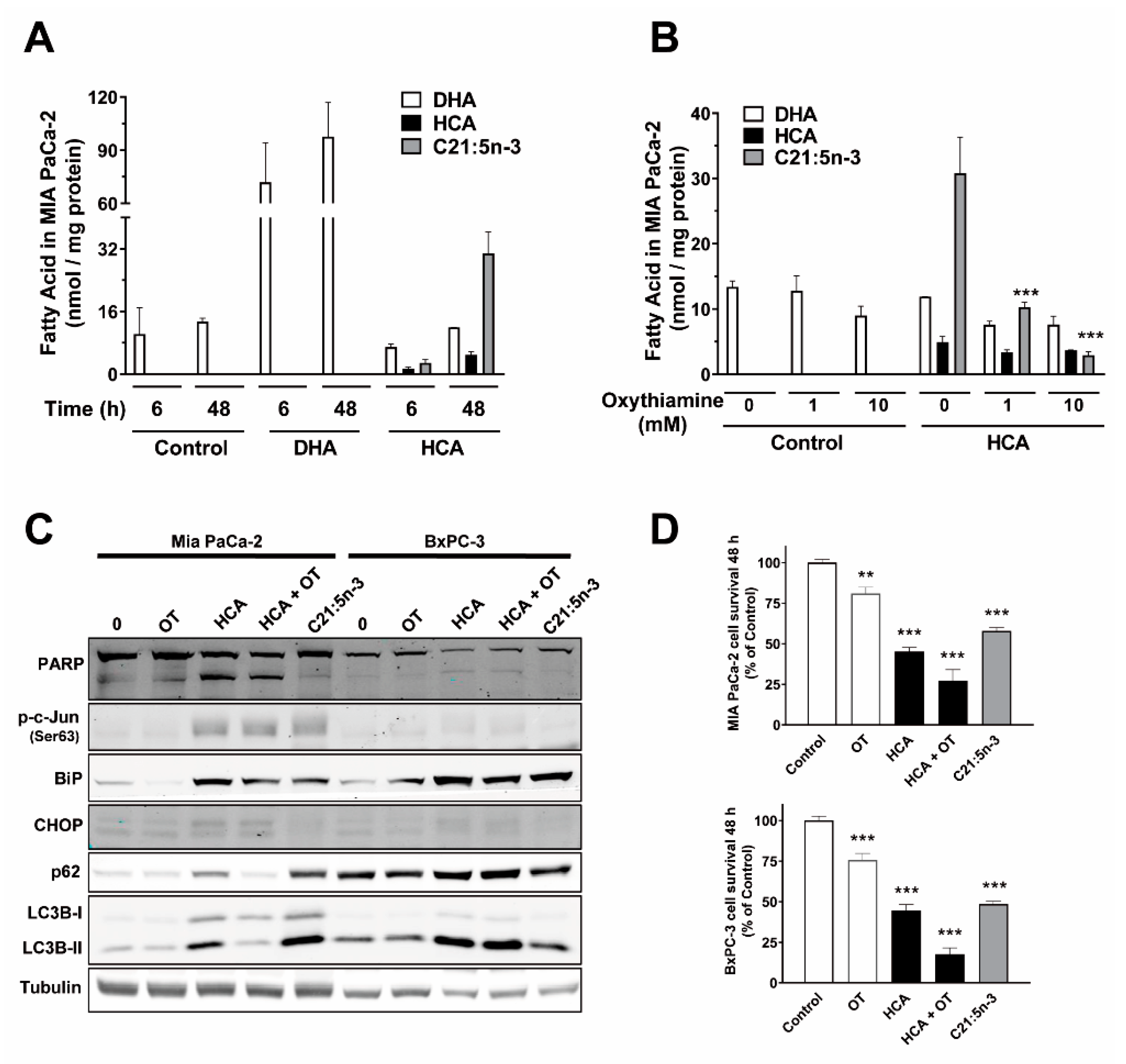
2.5. HCA Metabolization by α-Oxidation Does Not Play a Role in Its Mechanism of Action in Pancreatic Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures, Drugs, and Cell Proliferation Assays
4.2. Animals, Tumor Xenografts, and Treatments
4.3. Cell Cycle Analysis, Commercial Inhibitors, and Caspase-3/7 Activity
4.4. Cell Lysis, Protein Quantification, Electrophoresis, and Immunoblotting
4.5. Immunofluorescence and Confocal Microscopy
4.6. Fatty Acid Analysis by Gas Chromatography (GC)
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Goa, K.L. Gemcitabine. A review of its pharmacology and clinical potential in non-small cell lung cancer and pancreatic cancer. Drugs 1997, 54, 447–472. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Wee Ma, W.; Saleh, N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 18, 1691–1703. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Roth, M.T.; Cardin, D.B.; Berlin, J.D. Recent advances in the treatment of pancreatic cancer. F1000Research 2020, 9, 131. [Google Scholar] [CrossRef]
- Escribá, P.V. Membrane-lipid therapy: A new approach in molecular medicine. Trends Mol. Med. 2006, 12, 34–43. [Google Scholar] [CrossRef]
- Escribá, P.V. Membrane-lipid therapy: A historical perspective of membrane-targeted therapies—From lipid bilayer structure to the pathophysiological regulation of cells. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1493–1506. [Google Scholar] [CrossRef]
- Park, M.; Kim, H. Anti-cancer Mechanism of Docosahexaenoic Acid in Pancreatic Carcinogenesis: A Mini-review. J. Cancer Prev. 2017, 22, 1. [Google Scholar] [CrossRef] [Green Version]
- Beteta-Göbel, R.; Fernández-Díaz, J.; Arbona-González, L.; Rodríguez-Lorca, R.; Torres, M.; Busquets, X.; Fernández-García, P.; Escribá, P.V.; Lladó, V. The Novel Antitumor Compound HCA Promotes Glioma Cell Death by Inducing Endoplasmic Reticulum Stress and Autophagy. Cancers 2021, 13, 4290. [Google Scholar] [CrossRef]
- Torres, M.; Price, S.L.; Fiol-Deroque, M.A.; Marcilla-Etxenike, A.; Ahyayauch, H.; Barceló-Coblijn, G.; Terés, S.; Katsouri, L.; Ordinas, M.; López, D.J.; et al. Membrane lipid modifications and therapeutic effects mediated by hydroxydocosahexaenoic acid on Alzheimer’s disease. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1680–1692. [Google Scholar] [CrossRef]
- Fiol-Deroque, M.A.; Gutierrez-Lanza, R.; Terés, S.; Torres, M.; Barceló, P.; Rial, R.V.; Verkhratsky, A.; Escribá, P.V.; Busquets, X.; Rodríguez, J.J. Cognitive recovery and restoration of cell proliferation in the dentate gyrus in the 5XFAD transgenic mice model of Alzheimer’s disease following 2-hydroxy-DHA treatment. Biogerontology 2013, 14, 763–775. [Google Scholar] [CrossRef]
- Mohaibes, R.J.; Fiol-deRoque, M.A.; Torres, M.; Ordinas, M.; López, D.J.; Castro, J.A.; Escribá, P.V.; Busquets, X. The hydroxylated form of docosahexaenoic acid (DHA-H) modifies the brain lipid composition in a model of Alzheimer’s disease, improving behavioral motor function and survival. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1596–1603. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef]
- Fulda, S. Targeting apoptosis for anticancer therapy. Semin. Cancer Biol. 2015, 31, 84–88. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific Proteolytic Cleavage of Poly(ADP-ribose) Polymerase: An Early Marker of Chemotherapy-induced Apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526. [Google Scholar] [CrossRef]
- Shiomi, Y.; Yoshimura, M.; Kuki, K.; Hori, Y.; Tanaka, T. Z-360 Suppresses Tumor Growth in MIA PaCa-2-bearing Mice via Inhibition of Gastrin-induced Anti-Apoptotic Effects. Anticancer Res. 2017, 37, 4127–4137. [Google Scholar] [CrossRef]
- Westphal, S.; Kalthoff, H. Apoptosis: Targets in Pancreatic Cancer. Mol. Cancer 2003, 2, 6. [Google Scholar] [CrossRef]
- Parets, S.; Irigoyen, Á.; Ordinas, M.; Cabot, J.; Miralles, M.; Arbona, L.; Péter, M.; Balogh, G.; Fernández-García, P.; Busquets, X.; et al. 2-Hydroxy-Docosahexaenoic Acid Is Converted Into Heneicosapentaenoic Acid via α-Oxidation: Implications for Alzheimer’s Disease Therapy. Front. Cell Dev. Biol. 2020, 8, 164. [Google Scholar] [CrossRef]
- Guardiola-Serrano, F.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Ibarguren, M.; López, D.J.; Terés, S.; Alvarez, R.; Alonso-Sande, M.; Busquets, X.; Escribá, P.V. The novel anticancer drug hydroxytriolein inhibits lung cancer cell proliferation via a protein Kinase Cα- And extracellular signal-regulated kinase 1/2-Dependent Mechanism. J. Pharmacol. Exp. Ther. 2015, 354, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Foulon, V.; Sniekers, M.; Huysmans, E.; Asselberghs, S.; Mahieu, V.; Mannaerts, G.P.; Van Veldhoven, P.P.; Casteels, M. Breakdown of 2-hydroxylated straight chain fatty acids via peroxisomal 2-hydroxyphytanoyl-CoA lyase: A revised pathway for the α-oxidation of straight chain fatty acids. J. Biol. Chem. 2005, 280, 9802–9812. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef]
- Lu, H.; Lan, W.X.; Bo, L.; Niu, C.; Zhou, J.J.; Zhu, H.L. Metabolic response of LLC xenografted mice to oxythiamine, as measured by [1H] NMR spectroscopy. Genet. Mol. Res. 2015, 14, 11043–11051. [Google Scholar] [CrossRef]
- Irwin, S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia 1968, 13, 222–257. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Ali, S.M.; Khan, A.R.; Ahmad, M.U.; Chen, P.; Sheikh, S.; Ahmad, I. Synthesis and biological evaluation of gemcitabine–lipid conjugate (NEO6002). Bioorg. Med. Chem. Lett. 2005, 15, 2571–2574. [Google Scholar] [CrossRef]
- Hawryłkiewicz, A.; Ptaszyńska, N. Gemcitabine Peptide-Based Conjugates and Their Application in Targeted Tumor Therapy. Molecules 2021, 26, 364. [Google Scholar] [CrossRef]
- Torres, M.; Parets, S.; Fernández-Díaz, J.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Román, R.; Lladó, V.; Rosselló, C.A.; Fernández-García, P.; Escribá, P.V. Lipids in Pathophysiology and Development of the Membrane Lipid Therapy: New Bioactive Lipids. Membranes 2021, 11, 919. [Google Scholar] [CrossRef]
- Erazo, T.; Lorente, M.; López-Plana, A.; Munoz-Guardiola, P.; Ferńandez-Nogueira, P.; García-Martínez, J.A.; Bragado, P.; Fuster, G.; Salazar, M.; Espadaler, J.; et al. The New Antitumor Drug ABTL0812 Inhibits the Akt/mTORC1 Axis by Upregulating Tribbles-3 Pseudokinase. Clin. Cancer Res. 2016, 22, 2508–2519. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Na’Ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updat. 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Cremesti, A.; Paris, F.; Grassmé, H.; Holler, N.; Tschopp, J.; Fuks, Z.; Gulbins, E.; Kolesnick, R. Ceramide Enables Fas to Cap and Kill. J. Biol. Chem. 2001, 276, 23954–23961. [Google Scholar] [CrossRef]
- Nagesh, P.K.B.; Chowdhury, P.; Hatami, E.; Jain, S.; Dan, N.; Kashyap, V.K.; Chauhan, S.C.; Jaggi, M.; Yallapu, M.M. Tannic acid inhibits lipid metabolism and induce ROS in prostate cancer cells. Sci. Rep. 2020, 10, 980. [Google Scholar] [CrossRef]
- Wang, L.; Azad, N.; Kongkaneramit, L.; Chen, F.; Lu, Y.; Jiang, B.-H.; Rojanasakul, Y. The Fas Death Signaling Pathway Connecting Reactive Oxygen Species Generation and FLICE Inhibitory Protein Down-Regulation. J. Immunol. 2008, 180, 3072–3080. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Verfaillie, T.; Salazar, M.; Velasco, G.; Agostinis, P. Linking ER stress to autophagy: Potential implications for cancer therapy. Int. J. Cell Biol. 2010, 2010, 930509. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Lee, S.; Hong, E.; Jo, E.; Kim, Z.-H.; Yim, K.J.; Woo, S.H.; Choi, Y.-S.; Jang, H.-J. Gossypol Induces Apoptosis of Human Pancreatic Cancer Cells via CHOP/Endoplasmic Reticulum Stress Signaling Pathway. J. Microbiol. Biotechnol. 2022, 32, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef]
- Fernández-García, P.; Rosselló, C.A.; Rodríguez-Lorca, R.; Beteta-Göbel, R.; Fernández-Díaz, J.; Lladó, V.; Busquets, X.; Escribá, P.V. The opposing contribution of SMS1 and SMS2 to glioma progression and their value in the therapeutic response to 2OHOA. Cancers 2019, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Llado, V.; Gutierrez, A.; Martínez, J.; Casas, J.; Terés, S.; Higuera, M.; Galmés, A.; Saus, C.; Besalduch, J.; Busquets, X.; et al. Minerval induces apoptosis in Jurkat and other cancer cells. J. Cell. Mol. Med. 2010, 14, 659–670. [Google Scholar] [CrossRef]
- Ibarguren, M.; López, D.J.; Encinar, J.A.; González-Ros, J.M.; Busquets, X.; Escribá, P.V. Partitioning of liquid-ordered/liquid-disordered membrane microdomains induced by the fluidifying effect of 2-hydroxylated fatty acid derivatives. Biochim. Biophys. Acta 2013, 1828, 2553–2563. [Google Scholar] [CrossRef]
- Gajate, C.; Gayet, O.; Fraunhoffer, N.A.; Iovanna, J.; Dusetti, N.; Mollinedo, F. Induction of Apoptosis in Human Pancreatic Cancer Stem Cells by the Endoplasmic Reticulum-Targeted Alkylphospholipid Analog Edelfosine and Potentiation by Autophagy Inhibition. Cancers 2021, 13, 6124. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.; Ma, D.; Lee, W.-N.P.; Xiao, J.; Zhao, Y.; Go, V.L.; Wang, Q.; Yen, Y.; Recker, R.; et al. Inhibition of transketolase by oxythiamine altered dynamics of protein signals in pancreatic cancer cells. Exp. Hematol. Oncol. 2013, 2, 18. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, R.; Lee, W.N.P.; Deng, C.; Zhao, Y.; Lappe, J.; Recker, R.; Yen, Y.; Wang, Q.; Tsai, M.Y.; et al. Inhibition of protein phosphorylation in MIA pancreatic cancer cells: Confluence of metabolic and signaling pathways. J. Proteome Res. 2010, 9, 980. [Google Scholar] [CrossRef]
- Khalil, Y.; Carrino, S.; Lin, F.; Ferlin, A.; Lad, H.V.; Mazzacuva, F.; Falcone, S.; Rivers, N.; Banks, G.; Concas, D.; et al. Tissue Proteome of 2-Hydroxyacyl-CoA Lyase Deficient Mice Reveals Peroxisome Proliferation and Activation of ω-Oxidation. Int. J. Mol. Sci. 2022, 23, 987. [Google Scholar] [CrossRef]
- Mezzar, S.; De Schryver, E.; Asselberghs, S.; Meyhi, E.; Morvay, P.L.; Baes, M.; Van Veldhoven, P.P. Phytol-induced pathology in 2-hydroxyacyl-CoA lyase (HACL1) deficient mice. Evidence for a second non-HACL1-related lyase. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 972–990. [Google Scholar] [CrossRef]
- Guardiola-Serrano, F.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Ibarguren, M.; López, D.J.; Terés, S.; Alonso-Sande, M.; Higuera, M.; Torres, M.; Busquets, X.; et al. The triacylglycerol, hydroxytriolein, inhibits triple negative mammary breast cancer cell proliferation through a mechanism dependent on dihydroceramide and Akt. Oncotarget 2019, 10, 2486–2507. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Díaz, J.; Beteta-Göbel, R.; Torres, M.; Cabot, J.; Fernández-García, P.; Lladó, V.; Escribá, P.V.; Busquets, X. Tri-2-Hydroxyarachidonein Induces Cytocidal Autophagy in Pancreatic Ductal Adenocarcinoma Cancer Cell Models. Front. Physiol. 2022, 12, 2092. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beteta-Göbel, R.; Miralles, M.; Fernández-Díaz, J.; Rodríguez-Lorca, R.; Torres, M.; Fernández-García, P.; Escribá, P.V.; Lladó, V. HCA (2-Hydroxy-Docosahexaenoic Acid) Induces Apoptosis and Endoplasmic Reticulum Stress in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2022, 23, 9902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179902
Beteta-Göbel R, Miralles M, Fernández-Díaz J, Rodríguez-Lorca R, Torres M, Fernández-García P, Escribá PV, Lladó V. HCA (2-Hydroxy-Docosahexaenoic Acid) Induces Apoptosis and Endoplasmic Reticulum Stress in Pancreatic Cancer Cells. International Journal of Molecular Sciences. 2022; 23(17):9902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179902
Chicago/Turabian StyleBeteta-Göbel, Roberto, Marc Miralles, Javier Fernández-Díaz, Raquel Rodríguez-Lorca, Manuel Torres, Paula Fernández-García, Pablo V. Escribá, and Victoria Lladó. 2022. "HCA (2-Hydroxy-Docosahexaenoic Acid) Induces Apoptosis and Endoplasmic Reticulum Stress in Pancreatic Cancer Cells" International Journal of Molecular Sciences 23, no. 17: 9902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179902