
Unraveling the Role of the Tyrosine Tetrad from the Binding Site of the Epigenetic Writer MLL3 in the Catalytic Mechanism and Methylation Multiplicity
 , and
, and
Abstract
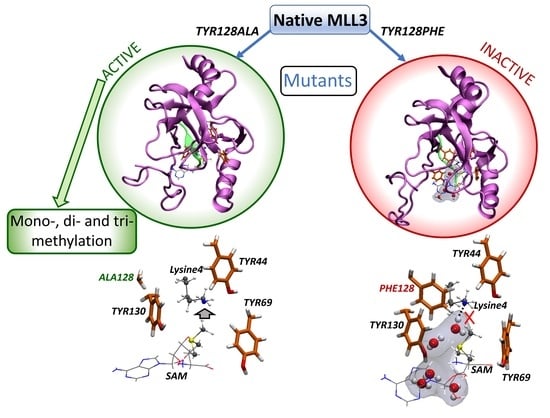
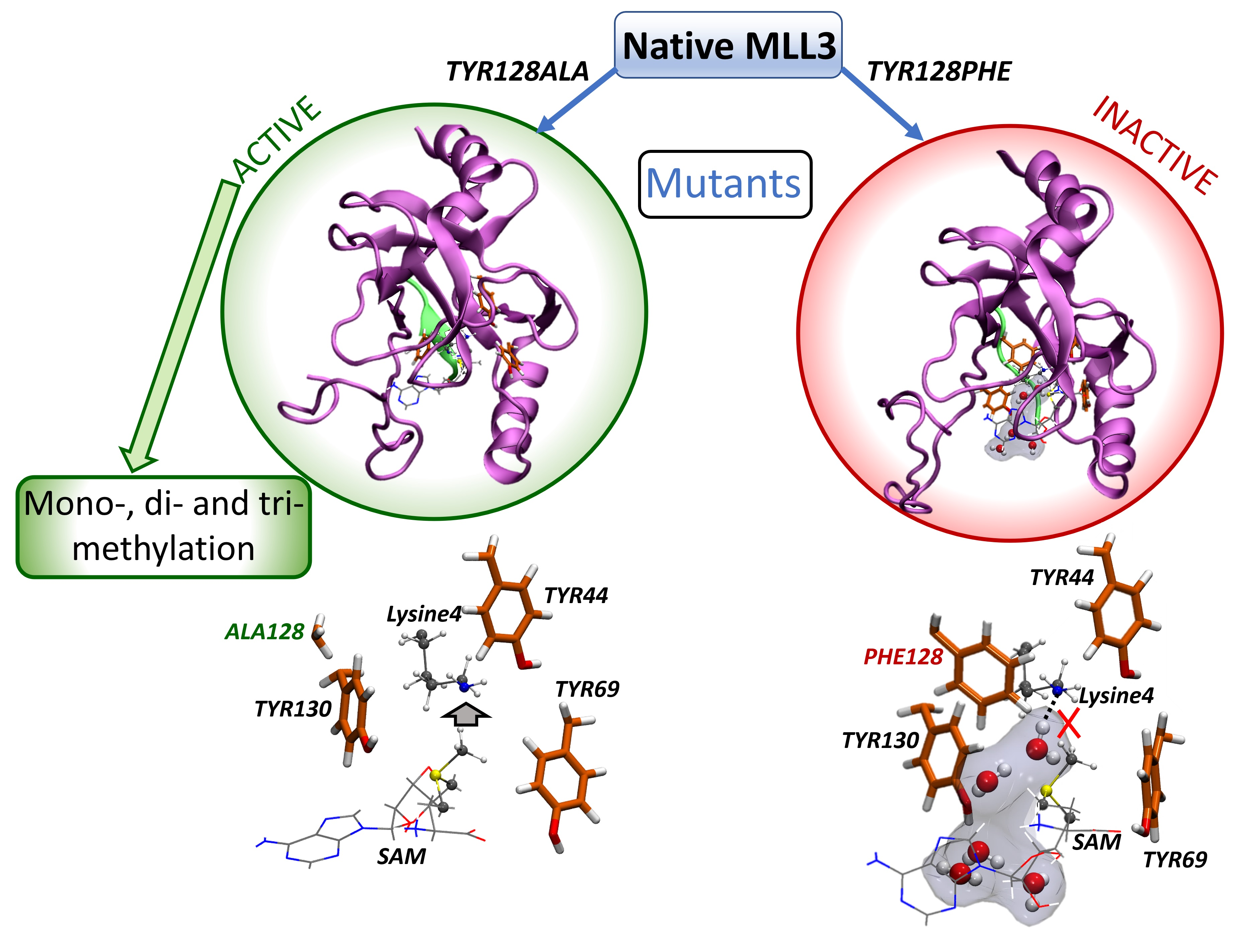
:
1. Introduction
2. Results and Discussion
2.1. Monomethyl-Transfer Reaction: Y44F/Y44A and Y128F/Y128A Mutants
2.2. Role of Y69F and Y130F Mutants in the Substrate-Inactivation Process
2.3. Di- and Trimethyl-Transfer Processes: The Effect of Mutations on Product Selectivity
2.4. Inactive Versus Active MLL3 Mutants
3. Materials and Methods
3.1. System Setup and Conformational Sampling
3.2. Setup of QM/MM Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002, 12, 198–209. [Google Scholar] [CrossRef]
- Bannister, A.J.; Schneider, R.; Kouzarides, T. Histone Methylation: Dynamic or Static? Cell 2002, 109, 801–806. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Farooq, Z.; Banday, S.; Pandita, T.K.; Altaf, M. The many faces of histone H3K79 methylation. Mutat. Res. Rev. Mutat. Res. 2016, 768, 46–52. [Google Scholar] [CrossRef]
- Evertts, A.G.; Manning, A.L.; Wang, X.; Dyson, N.J.; Garcia, B.A.; Coller, H.A. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 2013, 24, 3025–3037. [Google Scholar] [CrossRef]
- Murn, J.; Shi, Y. The winding path of protein methylation research: Milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18, 517–527. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Marques, S.; Outeiro, T.F. Epigenetics: Development and Disease. Epigenet. Dev. Dis. 2013, 61, 507–525. [Google Scholar] [CrossRef]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Jia, L.; Lv, W.; Wang, L.; Cui, W. Epigenetic enzyme mutations: Role in tumorigenesis and molecular inhibitors. Front. Oncol. 2019, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 Is a Haploinsufficient 7q Tumor Suppressor in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, D.-H.; Lee, S.; Yang, Q.-H.; Lee, D.K.; Lee, S.-K.; Roeder, R.G.; Lee, J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. USA 2009, 106, 8513–8518. [Google Scholar] [CrossRef]
- Wang, X.X.; Fu, L.; Li, X.; Wu, X.; Zhu, Z.; Fu, L.; Dong, J.T. Somatic mutations of the mixed-lineage leukemia 3 (MLL3) gene in primary breast cancers. Pathol. Oncol. Res. 2011, 17, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liu, H.Y.; Guo, S.H.; Sun, P.; Gong, F.M.; Jia, B.Q. Association of MLL3 expression with prognosis in gastric cancer. Genet. Mol. Res. 2014, 13, 7513–7518. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Steward, M.M.; Lee, J.S.; O’Donovan, A.; Wyatt, M.; Bernstein, B.E.; Shilatifard, A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 2006, 13, 852–854. [Google Scholar] [CrossRef]
- Xiao, B.; Jing, C.; Wilson, J.R.; Walker, P.A.; Vasisht, N.; Kelly, G.; Howell, S.; Taylor, I.A.; Blackburn, M.G.; Gamblin, S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652–656. [Google Scholar] [CrossRef]
- Li, Y.; Han, J.; Zhang, Y.; Cao, F.; Liu, Z.; Li, S.; Wu, J.; Hu, C.; Wang, Y.; Shuai, J.; et al. Structural basis for activity regulation of MLL family methyltransferases. Nature 2016, 530, 447–452. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Khan, S.I.; Horton, J.R.; Tamaru, H.; Selker, E.U.; Cheng, X. Structural basis for the product specificity of histone lysine methyltransferases. Mol. Cell 2003, 12, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Rojas, S.; Blanco-Esperguez, K.; Tuñón, I.; Kästner, J.; Mendizábal, F. Exploration of the Activation Mechanism of the Epigenetic Regulator MLL3: A QM/MM Study. Biomolecules 2021, 11, 1051. [Google Scholar] [CrossRef] [PubMed]
- Weirich, S.; Kudithipudi, S.; Kycia, I.; Jeltsch, A. Somatic cancer mutations in the MLL3-SET domain alter the catalytic properties of the enzyme. Clin. Epigenet. 2015, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.H.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Ruault, M.; Brun, M.E.; Ventura, M.; Roizès, G.; De Sario, A. MLL3, a new human member of the TRX/MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene 2002, 284, 73–81. [Google Scholar] [CrossRef]
- Van Der Meulen, J.; Sanghvi, V.; Mavrakis, K.; Durinck, K.; Fang, F.; Matthijssens, F.; Rondou, P.; Rosen, M.; Pieters, T.; Vandenberghe, P.; et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood 2015, 125, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Do Amaral Rabello, D.; De Moura, C.A.; De Andrade, R.V.; Motoyama, A.B.; Silva, F.P. Altered expression of MLL methyltransferase family genes in breast cancer. Int. J. Oncol. 2013, 43, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, D.; Chin, H.G.; Estève, P.-O.; Benner, J.; Jacobsen, S.E.; Pradhan, S. Substrate Specificity and Kinetic Mechanism of Mammalian G9a Histone H3 Methyltransferase. J. Biol. Chem. 2004, 279, 53248–53258. [Google Scholar] [CrossRef]
- Richardson, S.L.; Mao, Y.; Zhang, G.; Hanjra, P.; Peterson, D.L.; Huang, R. Kinetic Mechanism of Protein N-terminal Methyltransferase 1. J. Biol. Chem. 2015, 290, 11601–11610. [Google Scholar] [CrossRef] [Green Version]
- Collazo, E.; Couture, J.-F.; Bulfer, S.; Trievel, R.C. A coupled fluorescent assay for histone methyltransferases. Anal. Biochem. 2005, 342, 86–92. [Google Scholar] [CrossRef]
- Loring, H.S.; Thompson, P.R. Kinetic Mechanism of Nicotinamide N-Methyltransferase. Biochemistry 2018, 57, 5524–5532. [Google Scholar] [CrossRef]
- Kieseritzky, G.; Knapp, E.W. Improved pKa prediction: Combining empirical and semimicroscopic methods. J. Comput. Chem. 2008, 29, 2575–2581. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Senn, H.M.; O’Hagan, D.; Thiel, W. Insight into Enzymatic C–F Bond Formation from QM and QM/MM Calculations. J. Am. Chem. Soc. 2005, 127, 13643–13655. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Guo, H.-B.; Guo, H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. Proc. Natl. Acad. Sci. USA 2007, 104, 8797–8802. [Google Scholar] [CrossRef]
- Georgieva, P.; Himo, F. Quantum chemical modeling of enzymatic reactions: The case of histone lysine methyltransferase. J. Comput. Chem. 2010, 31, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Zhang, Y. Catalytic Mechanism and Product Specificity of the Histone Lysine Methyltransferase SET7/9: An ab Initio QM/MM-FE Study with Multiple Initial Structures. J. Am. Chem. Soc. 2006, 128, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-Optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Zhang, X.; Bruice, T.C. Histone Lysine Methyltransferase SET7/9: Formation of a Water Channel Precedes Each Methyl Transfer. Biochemistry 2007, 46, 14838–14844. [Google Scholar] [CrossRef]
- Kästner, J.; Sherwood, P. Superlinearly converging dimer method for transition state search. J. Chem. Phys. 2008, 128, 14106. [Google Scholar] [CrossRef]
- Metz, S.; Kästner, J.; Sokol, A.A.; Keal, T.W.; Sherwood, P. ChemShell—A modular software package for QM/MM simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 101–110. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | M3RA a | Y44F | Y128F | Y44A | Y128A | Y69F | Y130F |
|---|---|---|---|---|---|---|---|
| ΔE≠ | 18.9 ± 5.30 | 26.4 ± 9.9 | 37.7 ± 13.5 | 29.3 ± 9.6 | 20.3 ± 3.1 | 32.7 ± 16.6 | 34.8 ± 11.2 |
| ΔE° | −10.7 ± 7.6 | −14.2 ± 8.7 | −5.5 ± 7.2 | −5.4 ± 10.1 | −7.5 ± 5.1 | −5.4 ± 9.5 | −5.3 ± 9.5 |
| C-N(RS) | 3.19 ± 0.23 | 3.77 ± 0.61 | 4.16 ± 0.68 | 3.43 ± 0.37 | 3.19 ± 0.10 | 4.01 ± 1.11 | 3.74 ± 0.78 |
| C-N(TS) | 2.20 ± 0.05 | 2.28 ± 0.06 | 2.37 ± 0.14 | 2.21 ± 0.07 | 2.20 ± 0.05 | 2.25 ± 0.10 | 2.23 ± 0.07 |
| S-C(RS) | 1.82 ± 0.01 | 1.82 ± 0 | 1.81 ± 0.01 | 1.82 ± 0.01 | 1.81 ± 0.01 | 1.82 ± 0.01 | 1.81 ± 0.01 |
| S-C(TS) | 2.30 ± 0.04 | 2.34 ± 0.10 | 2.37 ± 0.12 | 2.41 ± 0.15 | 2.3 ± 0.11 | 2.44 ± 0.15 | 2.36 ± 0.13 |
| S-C-N(RS) | 151.0 ± 15.7 | 136 ± 19.4 | 123.8 ± 17.0 | 126.2 ± 26.4 | 132.8 ± 16.7 | 144.2 ± 19.0 | 116.2 ± 34.3 |
| S-C-N(TS) | 170.9 ± 4.5 | 166.7 ± 5.3 | 158.2 ± 12.6 | 161 ± 18.8 | 169.7 ± 3.5 | 167.9 ± 6.2 | 163.8 ± 8.0 |
| Systems | State | Y44-K4 a | Y128-K4 a | V68-Met b | R89-Met b | I91-Met b | Y130-Met c | Y69-SAM d |
|---|---|---|---|---|---|---|---|---|
| M3RA e | RS | 2.30 ± 0.34 | 2.55 ± 0.39 | 3.25 ± 1.10 | 3.20 ± 0.41 | 3.19 ± 0.45 | 3.11 ± 0.47 | 1.57 ± 0.03 |
| TS | 2.43 ± 0.49 | 2.48 ± 0.41 | 3.00 ± 1.06 | 3.22 ± 0.43 | 3.26 ± 0.34 | 2.93 ± 0.47 | 1.57 ± 0.03 | |
| Y44F | RS | --- | 2.31 ±0.42 | 2.66 ± 0.26 | 3.08 ± 0.63 | 3.77 ± 0.68 | 3.49 ± 0.75 | 4.73 ± 2.09 |
| TS | --- | 2.62 ± 0.71 | 2.65 ± 0.25 | 3.66 ± 0.58 | 3.43 ± 0.45 | 2.86 ± 0.50 | 4.65 ± 2.05 | |
| Y128F | RS | 2.36 ± 0.28 | --- | 2.60 ± 0.21 | 2.88 ± 0.49 | 3.25 ± 0.61 | 4.64 ± 1.22 | 2.78 ± 1.00 |
| TS | 2.44 ± 0.39 | --- | 2.59 ± 0.25 | 3.42 ± 0.51 | 3.04 ± 0.34 | 3.80 ± 1.05 | 2.68 ± 0.94 | |
| Y44A | RS | --- | 2.07 ± 0.36 | 2.53 ± 0.27 | 2.83 ± 0.38 | 3.31 ± 0.52 | 3.61 ± 0.71 | 1.99 ± 0.66 |
| TS | --- | 2.26 ± 0.26 | 2.46 ± 0.16 | 3.37 ± 0.81 | 3.29 ± 0.38 | 2.77 ± 0.50 | 1.97 ± 0.67 | |
| Y128A | RS | 2.57 ± 0.64 | --- | 2.87 ± 0.31 | 2.75 ± 0.35 | 3.14 ± 0.41 | 3.66 ± 0.52 | 1.70 ± 0.45 |
| TS | 2.44 ± 0.59 | --- | 2.54 ± 0.23 | 3.29 ± 0.40 | 3.05 ± 0.36 | 2.86 ± 0.39 | 1.69 ± 0.45 | |
| Y69F | RS | 3.16 ± 1.52 | 2.54 ± 1.19 | 3.39 ± 1.02 | 3.70 ± 1.15 | 3.93 ± 1.32 | 3.13 ± 0.57 | --- |
| TS | 3.71 ± 1.83 | 3.21 ± 1.26 | 2.96 ± 0.68 | 3.68 ± 0.97 | 3.68 ± 0.76 | 2.54 ± 0.26 | --- | |
| Y130F | RS | 3.32 ± 1.04 | 2.78 ± 0.94 | 3.00 ± 0.57 | 3.15 ± 0.80 | 3.84 ± 1.35 | --- | 2.28 ± 0.95 |
| TS | 3.40 ± 1.38 | 2.85 ± 0.94 | 2.61 ± 0.21 | 3.72 ± 0.82 | 3.55 ± 0.49 | --- | 2.13 ± 0.79 |
| Systems | Y44F-diMet | Y44A-diMet | Y128F-diMet | Y128A-diMet | Y44A-triMet | Y128A-triMet |
|---|---|---|---|---|---|---|
| ΔE≠ | 26.4 ± 7.2 | 15.5 ± 6.0 | 44.1 ± 16.7 | 15.8 ± 3.1 | 34.6 ± 11.7 | 15.2 ± 5.9 |
| ΔE° | −9.6 ± 5.4 | −14.9 ± 5.1 | 0.6 ± 8.9 | −12.9 ± 4.8 | −4.6 ± 7.0 | −17.3 ± 8.0 |
| C-N(RS) | 3.70 ± 0.60 | 3.16 ± 0.38 | 5.55 ± 0.75 | 3.14 ± 0.17 | 4.64 ± 0.83 | 3.25 ± 0.47 |
| C-N(TS) | 2.28 ± 0.05 | 2.22 ± 0.06 | 2.30 ± 0.08 | 2.21 ± 0.05 | 2.29 ± 0.09 | 2.26 ± 0.04 |
| S-C(RS) | 1.81 ± 0.01 | 1.82 ± 0.01 | 1.81 ± 0.01 | 1.82 ± 0.01 | 1.81 ± 0.01 | 1.81 ± 0.01 |
| S-C(TS) | 2.33 ± 0.08 | 2.28 ± 0.06 | 2.31 ± 0.09 | 2.29 ± 0.04 | 2.35 ± 0.10 | 2.28 ± 0.07 |
| S-C-N(RS) | 133.9 ± 28.2 | 155.3 ± 27.1 | 58.7 ± 12.9 | 151.7 ± 16.5 | 94.9 ± 32.6 | 137.4 ± 36.0 |
| S-C-N(TS) | 169.9 ± 6.0 | 171.5 ± 3.4 | 167.9 ± 5.0 | 171.3 ± 2.8 | 162.9 ± 19.1 | 171.8 ± 2.9 |
| Systems | State | Y44-K4 a | Y128-K4 a | V68-Met b | R89-Met b | I91-Met b | Y130-Met c | Y69-SAM d |
|---|---|---|---|---|---|---|---|---|
| Y44F-diMet | RS | --- | 2.36 ± 0.27 | 2.57 ± 0.25 | 2.55 ± 0.74 | 2.56 ± 0.88 | 2.55 ± 0.78 | 2.55 ± 1.19 |
| TS | --- | 2.45 ± 0.25 | 2.43 ± 0.12 | 2.40 ± 0.67 | 2.40 ± 0.40 | 2.39 ± 1.09 | 2.39 ± 1.23 | |
| Y44A-diMet | RS | --- | 2.65 ± 0.36 | 2.25 ± 1.42 | 3.17 ± 0.33 | 2.66 ± 0.69 | 2.95 ± 0.79 | 2.18 ± 1.27 |
| TS | --- | 2.71 ± 0.31 | 2.51 ± 0.13 | 3.22 ± 0.56 | 2.74 ± 0.43 | 2.72 ± 0.24 | 2.25 ± 1.42 | |
| Y128F-diMet | RS | 2.79 ± 0.37 | --- | 2.59 ± 0.42 | 3.32 ± 0.63 | 4.04 ± 0.39 | 2.63 ± 0.50 | 2.79 ± 0.59 |
| TS | 2.57 ± 0.20 | --- | 2.57 ± 0.40 | 3.47 ± 0.60 | 3.83 ± 0.44 | 3.06 ± 0.72 | 2.97 ± 0.71 | |
| Y128A-diMet | RS | 2.29 ± 0.12 | --- | 2.63 ± 0.26 | 2.95 ± 0.43 | 3.55 ± 0.51 | 3.31 ± 0.73 | 1.64 ± 0.21 |
| TS | 2.57 ± 0.29 | --- | 2.64 ± 0.38 | 2.79 ± 0.28 | 3.47 ± 0.46 | 2.90 ± 0.39 | 1.63 ± 0.21 | |
| Y44A-triMet | RS | --- | 3.94 ± 0.75 | 2.38 ± 0.10 | 3.77 ± 0.98 | 4.80 ± 1.68 | 5.18 ± 1.16 | 3.21 ± 1.32 |
| TS | --- | 4.03 ± 0.87 | 2.45 ± 0.18 | 3.65 ± 0.95 | 3.43 ± 0.67 | 3.36 ± 0.84 | 3.25 ± 1.32 | |
| Y128A-triMet | RS | 2.61 ± 0.17 | --- | 2.61 ± 0.29 | 3.27 ± 0.58 | 3.15 ± 1.13 | 4.32 ± 1.17 | 2.97 ± 1.56 |
| TS | 2.60 ± 0.17 | --- | 2.56 ± 0.36 | 3.57 ± 0.61 | 2.60 ± 0.31 | 3.31 ± 0.65 | 3.00 ± 1.44 |
| Mono-Methyl Transfer | ||||
|---|---|---|---|---|
| System | Y44F | Y44A | Y128F | Y128A |
| RS | 84 | 52 | 84 | 0 |
| TS | 12 | 0 | 0 | 0 |
| di-methyl transfer | ||||
| Y44F | Y44A | Y128F | Y128A | |
| RS | 72 | 16 | 60 | 8 |
| TS | 4 | 8 | 4 | 0 |
| tri-methyl transfer | ||||
| Y128A | Y44A | |||
| RS | 0 | 88 | ||
| TS | 0 | 0 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco-Esperguez, K.; Tuñón, I.; Kästner, J.; Mendizábal, F.; Miranda-Rojas, S. Unraveling the Role of the Tyrosine Tetrad from the Binding Site of the Epigenetic Writer MLL3 in the Catalytic Mechanism and Methylation Multiplicity. Int. J. Mol. Sci. 2022, 23, 10339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810339
Blanco-Esperguez K, Tuñón I, Kästner J, Mendizábal F, Miranda-Rojas S. Unraveling the Role of the Tyrosine Tetrad from the Binding Site of the Epigenetic Writer MLL3 in the Catalytic Mechanism and Methylation Multiplicity. International Journal of Molecular Sciences. 2022; 23(18):10339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810339
Chicago/Turabian StyleBlanco-Esperguez, Kevin, Iñaki Tuñón, Johannes Kästner, Fernando Mendizábal, and Sebastián Miranda-Rojas. 2022. "Unraveling the Role of the Tyrosine Tetrad from the Binding Site of the Epigenetic Writer MLL3 in the Catalytic Mechanism and Methylation Multiplicity" International Journal of Molecular Sciences 23, no. 18: 10339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810339