
Potent Chlorambucil-Platinum(IV) Prodrugs

 , and
, and 
Abstract
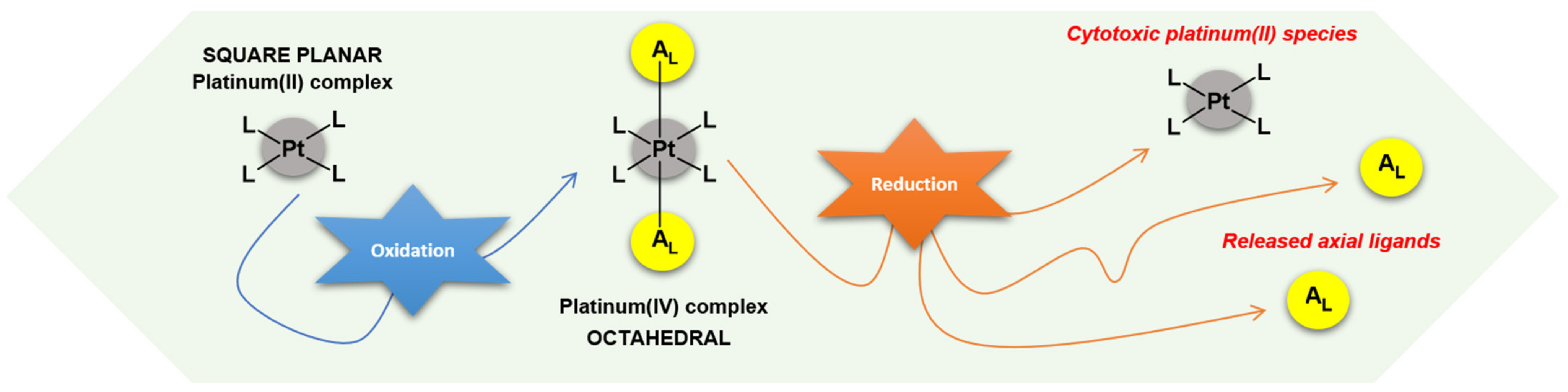
:1. Introduction
2. Results and Discussion
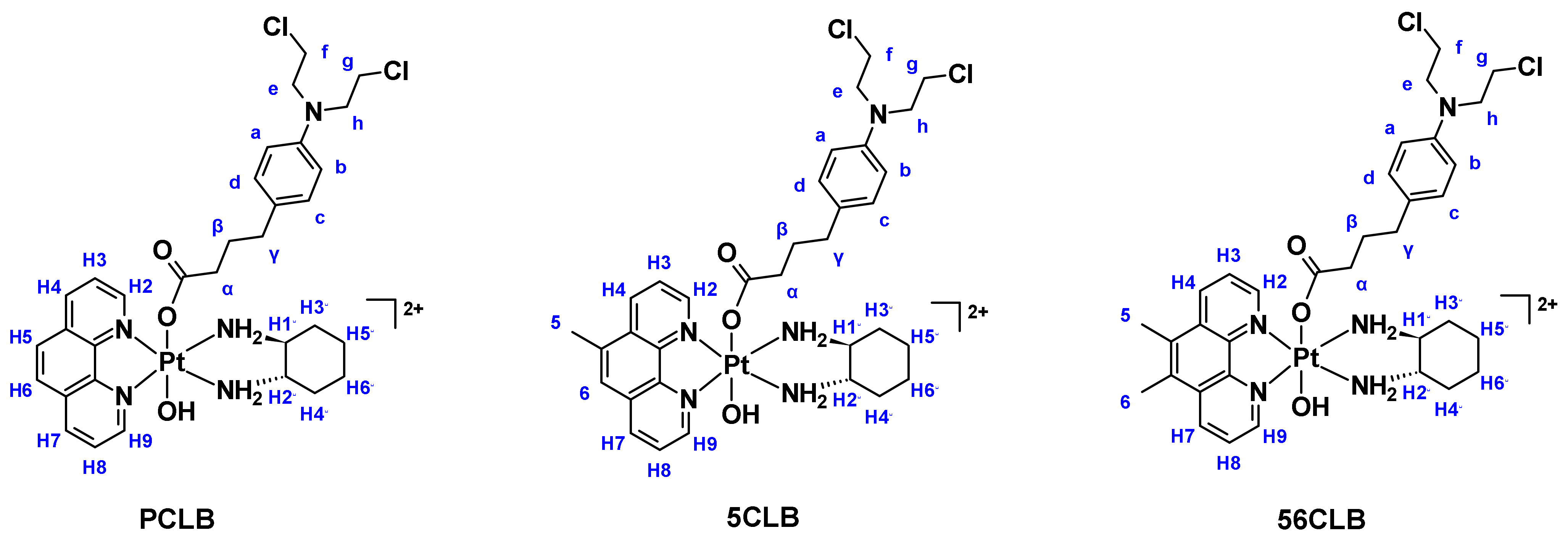
2.1. Synthesis and Characterization
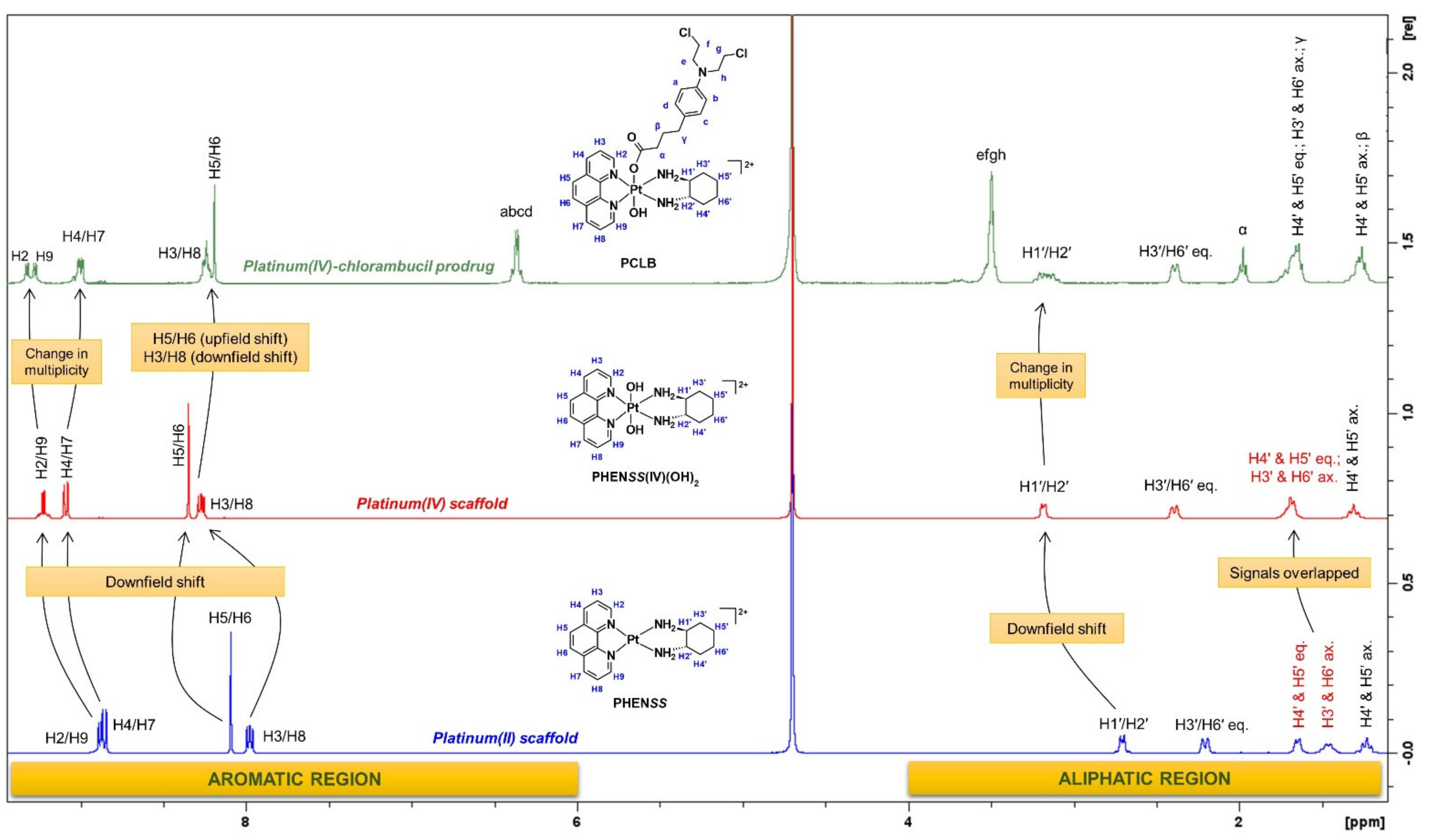
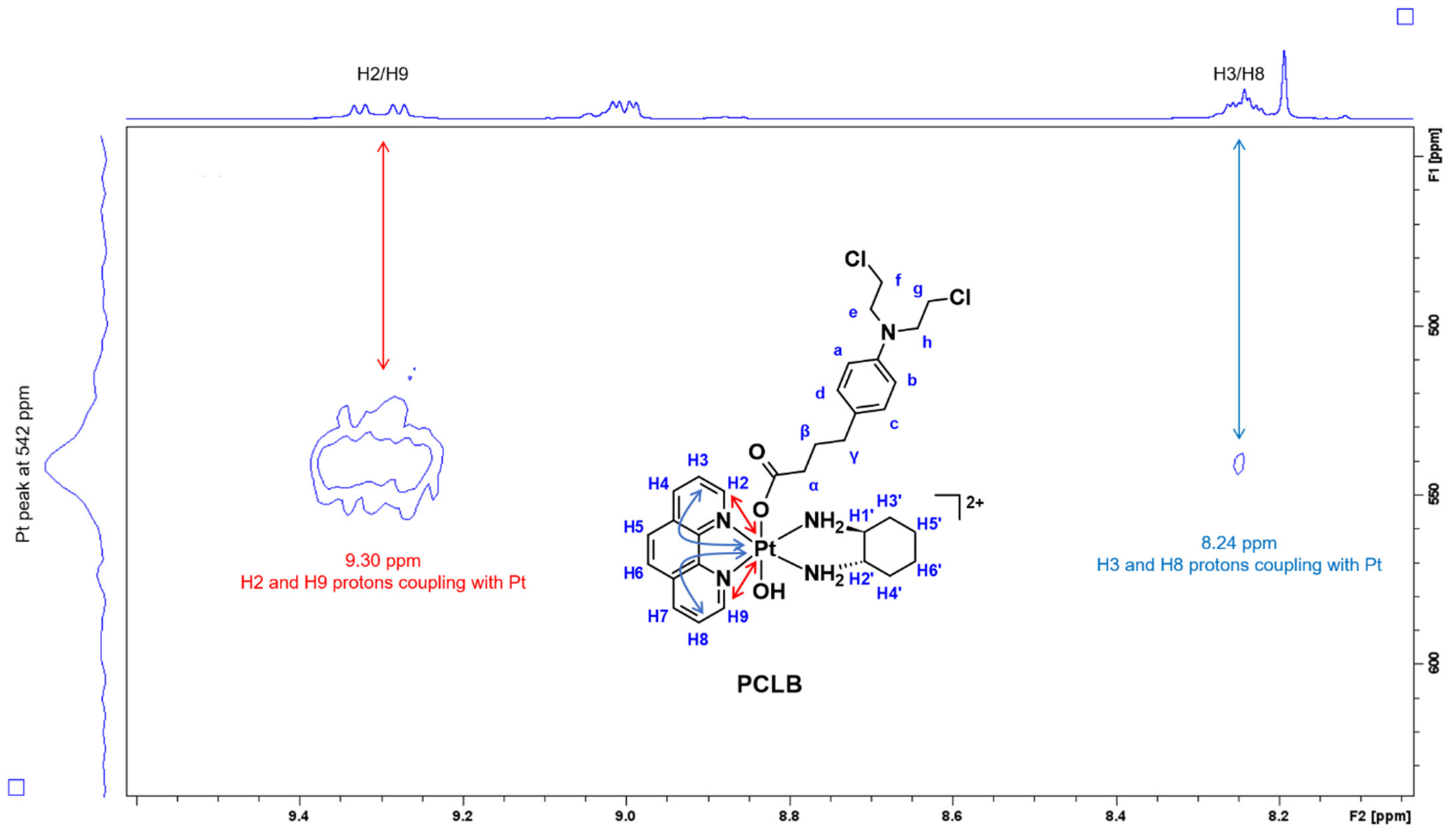
2.1.1. H-NMR and 1H-195Pt-HMQC Spectral Assignment
2.1.2. UV and CD Studies
2.2. Stability
2.3. Lipophilicity Studies
2.4. Reduction Experiments
2.5. Growth Inhibition Assays
2.6. ROS Production
3. Materials and Methods
3.1. Instrumentation
3.1.1. Flash Chromatography
3.1.2. High-Performance Liquid Chromatography (HPLC)
3.1.3. Nuclear Magnetic Resonance (NMR) Spectroscopy
3.1.4. Ultraviolet-Visible (UV) Spectroscopy
3.1.5. Circular Dichroism (CD) Spectroscopy
3.1.6. Electrospray Ionization Mass Spectrometry (ESI-MS)
3.2. Chemistry
3.2.1. General Synthesis of CLB Anhydride
3.2.2. General Synthesis of Precursor Platinum(II) Complexes of Type, [PtII(HL)(AL)]Cl2
3.2.3. General Synthesis of Precursor Platinum(IV) Dihydroxy Complexes of Type, [PtIV(HL)(AL)(OH)2](NO3)2
3.2.4. Synthesis of Platinum(IV) Complexes of Type, [PtIV(HL)(AL)(CLB)(OH)](NO3)2
3.3. Biological Investigations
3.3.1. Cell Growth Assays
3.3.2. Reactive Oxygen Species (ROS) Detection Assay
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siddik, Z.H.; Al-Baker, S.; Thai, G.; Khokhar, A.R. Antitumor activity of isomeric 1,2-diaminocyclohexane platinum(IV) complexes. J. Cancer Res. Clin. Oncol. 1994, 120, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L.R.; Sharp, S.Y.; O’Neill, C.F.; Raynaud, F.I.; Beale, P.J.; Judson, I.R. Mini-review: Discovery and development of platinum complexes designed to circumvent cisplatin resistance. J. Inorg. Biochem. 1999, 77, 111–115. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy-an update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef]
- Paprocka, R.; Wiese-Szadkowska, M.; Janciauskiene, S.; Kosmalski, T.; Kulik, M.; Helmin-Basa, A. Latest developments in metal complexes as anticancer agents. Coord. Chem. Rev. 2022, 452, 214307. [Google Scholar] [CrossRef]
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef]
- Wiltshaw, E. Cisplatin in the treatment of cancer-the first metal anti-tumour drug. Platin. Met. Rev. 1979, 23, 90–98. [Google Scholar]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Cleare, M.J.; Hoeschele, J.D. Studies on the antitumor activity of group VIII transition metal complexes. Part I. Platinum(II) complexes. Bioinorg. Chem. 1973, 2, 187–210. [Google Scholar] [CrossRef]
- Johnson, S.W.; Shen, D.W.; Pastan, I.; Gottesman, M.M.; Hamilton, T.C. Cross-resistance, cisplatin accumulation, and platinum–DNA adduct formation and removal in cisplatin-sensitive and -resistant human hepatoma cell lines. Exp. Cell Res. 1996, 226, 133–139. [Google Scholar] [CrossRef]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Kidera, Y.; Kawakami, H.; Sakiyama, T.; Okamoto, K.; Tanaka, K.; Takeda, M.; Kaneda, H.; Nishina, S.; Tsurutani, J.; Fujiwara, K.; et al. Risk factors for cisplatin-induced nephrotoxicity and potential of magnesium supplementation for renal protection. PLoS ONE 2014, 9, e101902–e101911. [Google Scholar] [CrossRef] [PubMed]
- Surendiran, A.; Balamurugan, N.; Gunaseelan, K.; Akhtar, S.; Reddy, K.S.; Adithan, C. Adverse drug reaction profile of cisplatin-based chemotherapy regimen in a tertiary care hospital in India: An evaluative study. Indian J. Pharmacol. 2010, 42, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Hartmann, J.T.; Fels, L.M.; Knop, S.; Stolt, H.; Kanz, L.; Bokemeyer, C. A randomized trial comparing the nephrotoxicity of cisplatin/ifosfamide-based combination chemotherapy with or without amifostine in patients with solid tumors. Investig. New Drugs 2000, 18, 281–289. [Google Scholar] [CrossRef]
- Coates, A.; Abraham, S.; Kaye, S.B.; Sowerbutts, T.; Frewin, C.; Fox, R.M.; Tattersall, M.H. On the receiving end–patient perception of the side-effects of cancer chemotherapy. Eur. J. Cancer Clin. Oncol. 1983, 19, 203–208. [Google Scholar] [CrossRef]
- Griffin, A.M.; Butow, P.N.; Coates, A.S.; Childs, A.M.; Ellis, P.M.; Dunn, S.M.; Tattersall, M.H. On the receiving end. V: Patient perceptions of the side effects of cancer chemotherapy in 1993. Ann. Oncol. 1996, 7, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, J.; Boursiquot, J.N.; Cournoyer, G.; Lemieux, J.; Masse, M.S.; Almanric, K.; Guay, M.P.; Comite de l’evolution des pratiques en o. Management of hypersensitivity to platinum- and taxane-based chemotherapy: Cepo review and clinical recommendations. Curr. Oncol. 2014, 21, 630–641. [Google Scholar] [CrossRef]
- Cameron, A.C.; Touyz, R.M.; Lang, N.N. Vascular complications of cancer chemotherapy. Can. J. Cardiol. 2016, 32, 852–862. [Google Scholar] [CrossRef]
- Tsang, R.Y.; Al-Fayea, T.; Au, H.J. Cisplatin overdose: Toxicities and management. Drug Saf. 2009, 32, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Galanski, M.; Jakupec, M.A.; Keppler, B.K. Update of the preclinical situation of anticancer platinum complexes: Novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Busselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Bugarcic, Z.D.; Bogojeski, J.; Petrovic, B.; Hochreuther, S.; van Eldik, R. Mechanistic studies on the reactions of platinum(II) complexes with nitrogen- and sulfur-donor biomolecules. Dalton Trans. 2012, 41, 12329–12345. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R. Interactions of cisplatin with non-DNA targets and their influence on anticancer activity and drug toxicity: The complex world of the platinum complex. Curr. Cancer Drug Targets 2015, 14, 794–816. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Hambley, T.W. Platinum(IV) antitumour compounds: Their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar] [CrossRef]
- Wong, D.Y.Q.; Ang, W.H. Development of platinum(IV) complexes as anticancer prodrugs: The story so far. COSMOS 2012, 8, 121–135. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, Z.; Zhu, G. Emerging platinum(IV) prodrugs to combat cisplatin resistance: From isolated cancer cells to tumor microenvironment. Dalton Trans. 2019, 48, 2536–2544. [Google Scholar] [CrossRef]
- Jia, C.; Deacon, G.B.; Zhang, Y.; Gao, C. Platinum(IV) antitumor complexes and their nano-drug delivery. Coord. Chem. Rev. 2021, 429, 213640–213675. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Deng, Z.; Zhu, G. Recent advances in the synthesis, stability, and activation of platinum(IV) anticancer prodrugs. Coord. Chem. Rev. 2021, 442, 213991–214023. [Google Scholar] [CrossRef]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. Pt(IV) antitumor prodrugs: Dogmas, paradigms, and realities. Dalton Trans. 2022, 51, 2121–2134. [Google Scholar] [CrossRef] [PubMed]
- Basu, U.; Banik, B.; Wen, R.; Pathak, R.K.; Dhar, S. The Platin-X series: Activation, targeting, and delivery. Dalton Trans. 2016, 45, 12992–13004. [Google Scholar] [CrossRef] [PubMed]
- Wexselblatt, E.; Gibson, D. What do we know about the reduction of Pt(IV) pro-drugs? J. Inorg. Biochem. 2012, 117, 220–229. [Google Scholar] [CrossRef]
- Latosińska, J.; Latosińska, M. Anticancer Drug Discovery—From Serendipity to Rational Design; IntechOpen: London, UK, 2013. [Google Scholar]
- Bhargava, A.; Vaishampayan, U.N. Satraplatin: Leading the new generation of oral platinum agents. Expert Opin. Investig. Drugs 2009, 18, 1787–1797. [Google Scholar] [CrossRef]
- Bell, D.N.; Liu, J.J.; Tingle, M.D.; Rattel, B.; Meyer, T.U.; McKeage, M.J. Comparative protein binding, stability and degradation of satraplatin, JM118 and cisplatin in human plasma in vitro. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1440–1446. [Google Scholar]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. A view on multi-action Pt(IV) antitumor prodrugs. Inorg. Chim. Acta 2019, 492, 32–47. [Google Scholar] [CrossRef]
- Brodie, C.R.; Collins, J.G.; Aldrich-Wright, J.R. DNA binding and biological activity of some platinum(II) intercalating compounds containing methyl-substituted 1,10-phenanthrolines. Dalton Trans. 2004, 8, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Taleb, R.I.; Krause-Heuer, A.M.; Cook, R.L.; Wang, S.; Higgins, V.J.; Aldrich-Wright, J.R. Novel platinum(II)-based anticancer complexes and molecular hosts as their drug delivery vehicles. Dalton Trans. 2007, 43, 5055–5064. [Google Scholar] [CrossRef]
- Fisher, D.M.; Bednarski, P.J.; Grünert, R.; Turner, P.; Fenton, R.R.; Aldrich-Wright, J.R. Chiral platinum(II) metallointercalators with potent in vitro cytotoxic activity. ChemMedChem 2007, 2, 488–495. [Google Scholar] [CrossRef]
- Kemp, S.; Wheate, N.J.; Buck, D.P.; Nikac, M.; Collins, J.G.; Aldrich-Wright, J.R. The effect of ancillary ligand chirality and phenanthroline functional group substitution on the cytotoxicity of platinum(II)-based metallointercalators. J. Inorg. Biochem. 2007, 101, 1049–1058. [Google Scholar] [CrossRef]
- Krause-Heuer, A.M.; Grunert, R.; Kuhne, S.; Buczkowska, M.; Wheate, N.J.; Le Pevelen, D.D.; Boag, L.R.; Fisher, D.M.; Kasparkova, J.; Malina, J.; et al. Studies of the mechanism of action of platinum(II) complexes with potent cytotoxicity in human cancer cells. J. Med. Chem. 2009, 52, 5474–5484. [Google Scholar] [CrossRef] [PubMed]
- Kostrhunova, H.; Zajac, J.; Novohradsky, V.; Kasparkova, J.; Malina, J.; Aldrich-Wright, J.R.; Petruzzella, E.; Sirota, R.; Gibson, D.; Brabec, V. A subset of new platinum antitumor agents kills cells by a multimodal mechanism of action also involving changes in the organization of the microtubule cytoskeleton. J. Med. Chem. 2019, 62, 5176–5190. [Google Scholar] [CrossRef] [PubMed]
- Garbutcheon-Singh, K.B.; Myers, S.; Harper, B.W.; Ng, N.S.; Dong, Q.; Xie, C.; Aldrich-Wright, J.R. The effects of 56MESS on mitochondrial and cytoskeletal proteins and the cell cycle in MDCK cells. Metallomics 2013, 5, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.B. Metabolism and reactions of alkylating agents. Pharmacol. Ther. 1987, 35, 301–358. [Google Scholar] [CrossRef]
- Di Antonio, M.; McLuckie, K.I.E.; Balasubramanian, S. Reprogramming the mechanism of action of chlorambucil by coupling to a G-quadruplex ligand. J. Am. Chem. Soc. 2014, 136, 5860–5863. [Google Scholar] [CrossRef]
- Löf, K.; Hovinen, J.; Reinikainen, P.; Vilpo, L.M.; Seppälä, E.; Vilpo, J.A. Kinetics of chlorambucil in vitro: Effects of fluid matrix, human gastric juice, plasma proteins and red cells. Chem. Biol. Interact. 1997, 103, 187–198. [Google Scholar] [CrossRef]
- Ichida, F.; Konishi, T.; Asada, R.; Yamatani, M.; Konda, M.; Tani, M.; Tanizawa, T.; Suzuki, Y.; Okada, T.; Kyotani, S.; et al. Chlorambucil central nervous toxicity: A significant side effect of chlorambucil therapy in childhood nephrotic syndrome. Eur. J. Pediatr. 1985, 144, 283–286. [Google Scholar] [CrossRef]
- Peterman, A.; Braunstein, B. Cutaneous reaction to chlorambucil therapy. Arch. Dermatol. 1986, 122, 1358–1360. [Google Scholar] [CrossRef]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione transferases: Potential targets to overcome chemoresistance in solid tumors. Int. J. Mol. Sci. 2018, 19, 3785. [Google Scholar] [CrossRef]
- Ma, Z.-Y.; Wang, D.-B.; Song, X.-Q.; Wu, Y.-G.; Chen, Q.; Zhao, C.-L.; Li, J.-Y.; Cheng, S.-H.; Xu, J.-Y. Chlorambucil-conjugated platinum(IV) prodrugs to treat triple-negative breast cancer in vitro and in vivo. Eur. J. Med. Chem. 2018, 157, 1292–1299. [Google Scholar] [CrossRef]
- Qin, X.; Fang, L.; Chen, F.; Gou, S. Conjugation of platinum(IV) complexes with chlorambucil to overcome cisplatin resistance via a “joint action” mode toward DNA. Eur. J. Med. Chem. 2017, 137, 167–175. [Google Scholar] [CrossRef]
- Montagner, D.; Tolan, D.; Andriollo, E.; Gandin, V.; Marzano, C. A Pt(IV) prodrug combining chlorambucil and cisplatin: A dual-acting weapon for targeting DNA in cancer cells. Int. J. Mol. Sci. 2018, 19, 3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, R.K.; Wen, R.; Kolishetti, N.; Dhar, S. A prodrug of two approved drugs, cisplatin and chlorambucil, for chemo war against cancer. Mol. Cancer Ther. 2017, 16, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Petruzzella, E.; Braude, J.P.; Aldrich-Wright, J.R.; Gandin, V.; Gibson, D. A quadruple-action platinum(IV) prodrug with anticancer activity against KRAS mutated cancer cell lines. Angew. Chem. Int. Ed. Engl. 2017, 56, 11539–11544. [Google Scholar] [CrossRef] [PubMed]
- Harper, B.W.J.; Petruzzella, E.; Sirota, R.; Faccioli, F.F.; Aldrich-Wright, J.R.; Gandin, V.; Gibson, D. Synthesis, characterization and in vitro and in vivo anticancer activity of Pt(iv) derivatives of [Pt(1S,2S-DACH)(5,6-dimethyl-1,10-phenanthroline)]. Dalton Trans. 2017, 46, 7005–7019. [Google Scholar] [CrossRef]
- Aputen, A. Novel Platinum(IV) Prodrugs. Master’s Thesis, Western Sydney University, Campbelltown, Australia, 2019. [Google Scholar]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Aldrich Wright, J.R. Synthesis, characterisation and potent cytotoxicity of unconventional platinum(IV) complexes with modified lipophilicity. Dalton Trans. 2019, 48, 17217–17227. [Google Scholar] [CrossRef]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Aldrich Wright, J.R. Synthesis, characterisation and influence of lipophilicity on cellular accumulation and cytotoxicity of unconventional platinum(iv) prodrugs as potent anticancer agents. Dalton Trans. 2019, 48, 17228–17240. [Google Scholar] [CrossRef]
- Macias, F.J.; Deo, K.M.; Pages, B.J.; Wormell, P.; Clegg, J.K.; Zhang, Y.; Li, F.; Zheng, G.; Sakoff, J.; Gilbert, J.; et al. Synthesis and analysis of the structure, diffusion and cytotoxicity of heterocyclic platinum(IV) complexes. Chem. Eur. J. 2015, 21, 16990–17001. [Google Scholar] [CrossRef]
- Khoury, A.; Sakoff, J.A.; Gilbert, J.; Scott, K.F.; Karan, S.; Gordon, C.P.; Aldrich-Wright, J.R. Cyclooxygenase-inhibiting platinum(iv) prodrugs with potent anticancer activity. Pharmaceutics 2022, 14, 787. [Google Scholar] [CrossRef]
- Garbutcheon-Singh, K.B.; Leverett, P.; Myers, S.; Aldrich-Wright, J.R. Cytotoxic platinum(II) intercalators that incorporate 1R,2R-diaminocyclopentane. Dalton Trans. 2013, 42, 918–926. [Google Scholar] [CrossRef]
- McGhie, B.S.; Sakoff, J.; Gilbert, J.; Aldrich-Wright, J.R. Synthesis and characterisation of platinum(IV) polypyridyl complexes with halide axial ligands. Inorg. Chim. Acta 2019, 495, 118964–118974. [Google Scholar] [CrossRef]
- Pages, B.J.; Li, F.; Wormell, P.; Ang, D.L.; Clegg, J.K.; Kepert, C.J.; Spare, L.K.; Danchaiwijit, S.; Aldrich-Wright, J.R. Synthesis and analysis of the anticancer activity of platinum(II) complexes incorporating dipyridoquinoxaline variants. Dalton Trans. 2014, 43, 15566–15575. [Google Scholar] [CrossRef] [Green Version]
- Owen, W.R.; Stewart, P.J. Kinetics and mechanism of chlorambucil hydrolysis. J. Pharm. Sci. 1979, 68, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Valkó, K.L. Lipophilicity and biomimetic properties measured by HPLC to support drug discovery. J. Pharm. Biomed. Anal. 2016, 130, 35–54. [Google Scholar] [CrossRef]
- Klose, M.H.M.; Theiner, S.; Varbanov, H.P.; Hoefer, D.; Pichler, V.; Galanski, M.; Meier-Menches, S.M.; Keppler, B.K. Development and validation of liquid chromatography-based methods to assess the lipophilicity of cytotoxic platinum(iv) complexes. Inorganics 2018, 6, 130. [Google Scholar] [CrossRef]
- Gabano, E.; Ravera, M.; Perin, E.; Zanellato, I.; Rangone, B.; McGlinchey, M.J.; Osella, D. Synthesis and characterization of cyclohexane-1R,2R-diamine-based Pt(IV) dicarboxylato anticancer prodrugs: Their selective activity against human colon cancer cell lines. Dalton Trans. 2019, 48, 435–445. [Google Scholar] [CrossRef]
- Valkó, K. Application of high-performance liquid chromatography based measurements of lipophilicity to model biological distribution. J. Chromatogr. A 2004, 1037, 299–310. [Google Scholar] [CrossRef]
- Chen, C.K.J.; Gui, X.; Kappen, P.; Renfrew, A.K.; Hambley, T.W. The effect of charge on the uptake and resistance to reduction of platinum(IV) complexes in human serum and whole blood models. Metallomics 2020, 12, 1599–1615. [Google Scholar] [CrossRef]
- Chen, C.K.J.; Kappen, P.; Gibson, D.; Hambley, T.W. Trans-platinum(IV) pro-drugs that exhibit unusual resistance to reduction by endogenous reductants and blood serum but are rapidly activated inside cells: 1H NMR and XANES spectroscopy study. Dalton Trans. 2020, 49, 7722–7736. [Google Scholar] [CrossRef]
- Chen, C.K.J.; Kappen, P.; Hambley, T.W. The reduction of cis-platinum(IV) complexes by ascorbate and in whole human blood models using 1H NMR and XANES spectroscopy. Metallomics 2019, 11, 686–695. [Google Scholar] [CrossRef]
- Choi, S.; Filotto, C.; Bisanzo, M.; Delaney, S.; Lagasee, D.; Whitworth, J.L.; Jusko, A.; Li, C.; Wood, N.A.; Willingham, J.; et al. Reduction and anticancer activity of platinum(IV) complexes. Inorg. Chem. 1998, 37, 2500–2504. [Google Scholar] [CrossRef]
- Deo, K.M. Synthesis, Characterization, and Reduction Studies of Novel Platinum(IV) Metallointercalators. Bachelor’s Thesis, Western Sydney University, Campbelltown, Australia, 2014. [Google Scholar]
- Tolan, D.; Gandin, V.; Morrison, L.; El-Nahas, A.; Marzano, C.; Montagner, D.; Erxleben, A. Oxidative stress induced by Pt(IV) pro-drugs based on the cisplatin scaffold and indole carboxylic acids in axial position. Sci. Rep. 2016, 6, 29367–29379. [Google Scholar] [CrossRef] [PubMed]
- Harper, B.W.; Friedman-Ezra, A.; Sirota, R.; Petruzzella, E.; Aldrich-Wright, J.R.; Gibson, D. Probing the interactions of cytotoxic [Pt(1S,2S-DACH)(5,6-dimethyl-1,10-phenanthroline)] and its Pt(IV) derivatives with human serum. ChemMedChem 2017, 12, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Moretto, J.; Chauffert, B.; Ghiringhelli, F.; Aldrich-Wright, J.R.; Bouyer, F. Discrepancy between in vitro and in vivo antitumor effect of a new platinum(II) metallointercalator. Investig. New Drugs 2011, 29, 1164–1176. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084–101092. [Google Scholar] [CrossRef]
- Zhuang, J.; Liu, Y.; Yuan, Q.; Liu, J.; Liu, Y.; Li, H.; Wang, D. Blue light-induced apoptosis of human promyelocytic leukemia cells via the mitochondrial-mediated signaling pathway. Oncol. Lett. 2018, 15, 6291–6297. [Google Scholar] [CrossRef]
- Oh, P.-S.; Hwang, H.; Jeong, H.-S.; Kwon, J.; Kim, H.-S.; Kim, M.; Lim, S.; Sohn, M.-H.; Jeong, H.-J. Blue light emitting diode induces apoptosis in lymphoid cells by stimulating autophagy. Int. J. Biochem. Cell Biol. 2016, 70, 13–22. [Google Scholar] [CrossRef]
- Oh, P.-S.; Na, K.S.; Hwang, H.; Jeong, H.-S.; Lim, S.; Sohn, M.-H.; Jeong, H.-J. Effect of blue light emitting diodes on melanoma cells: Involvement of apoptotic signaling. J. Photochem. Photobiol. B Biol. 2015, 142, 197–203. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Tarleton, M.; Gilbert, J.; Robertson, M.J.; McCluskey, A.; Sakoff, J.A. Library synthesis and cytotoxicity of a family of 2-phenylacrylonitriles and discovery of an estrogen dependent breast cancer lead compound. MedChemComm 2011, 2, 31–37. [Google Scholar] [CrossRef]
- Fayad, C.; Audi, H.; Khnayzer, R.S.; Daher, C.F. The anti-cancer effect of series of strained photoactivatable Ru(II) polypyridyl complexes on non-small-cell lung cancer and triple negative breast cancer cells. J. Biol. Inorg. Chem. 2020, 26, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.G.; Mehanna, S.; Elias, E.; Khnayzer, R.S.; Daher, C.F. A photoactivatable chemotherapeutic Ru(II) complex bearing bathocuproine ligand efficiently induces cell death in human malignant melanoma cells through a multi-mechanistic pathway. Chem. Bio. Interact. 2021, 348, 109644. [Google Scholar] [CrossRef]
- Mehanna, S.; Mansour, N.; Daher, C.F.; Elias, M.G.; Dagher, C.; Khnayzer, R.S. Drug-free phototherapy of superficial tumors: White light at the end of the tunnel. J. Photochem. Photobiol. B Biol. 2021, 224, 112324–112335. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proton Labels | PCLB | 5CLB | 56CLB |
|---|---|---|---|
| H2 | 9.32 (d, 1H, J = 5.5 Hz) | 9.26 (qd, 2H) | 9.25 (d, 1H, J = 5.4 Hz) |
| H9 | 9.27 (d, 1H, J = 5.5 Hz) | 9.22 (d, 1H, J = 5.4 Hz) | |
| H4 | 8.99 (dd, 2H, J1 = 8.3, J2 = 3.3 Hz) | 9.05 (dd, 1H, J1 = 8.4, J2 = 4.3 Hz) | 9.10 (dd, 2H, J1 = 8.6, J2 = 5 Hz) |
| H7 | 8.87 (dd, 1H, J1 = 8.3, J2 = 3.6 Hz) | ||
| H3 | 8.24 (m, 2H) | 8.26 (m, 1H) | 8.23 (q, 2H, J = 5.5 Hz) |
| H8 | 8.19 (m, 1H) | ||
| H5 and H6 | 8.20 (s, 2H) | - | - |
| H6 | - | 7.95 (d, 1H, J = 4.5 Hz) | - |
| a and b | 6.37 (q, 4H, J = 8.8 Hz) (resonances overlapped) | 6.27 (q, 4H, J = 8.3 Hz) (slight distance between a and b and c and d resonances) | 6.24 (q, 4H, J = 8.6 Hz) (greater distance between a and b and c and d resonances) |
| c and d | |||
| e, f, g, and h | 3.51 (m, 8H) (sharp resonance) | 3.48 (s, 8H) | 3.52 (s, 8H) |
| H1’ and H2’ | 3.17 (m, 2H) | 3.16 (m, 2H) | 3.17 (m, 2H) |
| CH3(5 positions) | - | 2.72 (s, 3H) | - |
| CH3(5 and 6 positions) | - | - | 2.63 (d, 6H, J = 3.6 Hz) |
| H3’ and H6’ eq. | 2.38 (d, 2H) | 2.38 (d, 2H) | 2.39 (d, 2H) |
| α | 1.97 (t, 2H) | 1.97 (t, 2H) | 1.97 (t, 2H) |
| β | Overlapping with H4’ and H5’ ax. | Overlapping with H4’ and H5’ ax. | Overlapping with H4’ and H5’ ax. |
| γ | Overlapping with H4’ and H5’ eq.; H3’ and H6’ ax. | 1.46 (m, 2H) | Overlapping with H4’ and H5’ ax. |
| H4’ and H5’ eq.; H3’ and H6’ ax. | Overlapping with γ | 1.68 (m, 4H) | - |
| H4’ and H5’ ax. | Overlapping with β | Overlapping with β | - |
| H4’ and H5’ eq.; H3’ and H6’ ax. | - | 1.68 (m, 4H) | 1.69 (m, 4H) |
| H4’ and H5’ eq.; H3’ and H6’ ax.; γ | 1.68 (m, 6H) | - | - |
| H4’ and H5’ ax.; β | 1.27 (m, 4H) | 1.25 (m, 4H) | - |
| H4’ and H5’ ax.; β; γ | - | - | 1.20 (m, 6H) |
| 1H/195Pt | 9.30, 8.24/542 ppm | 9.26/544 ppm | 9.25, 9.22, 8.23/532 ppm |
| Platinum(IV)-CLB Complexes | UV λmax nm (ε/M.cm−1 ± SD × 104) | CD λmax nm (Δε/M.cm−1 × 101) |
|---|---|---|
| PCLB | 279 (2.92 ± 0.82), 203 (9.48 ± 2.20) | 206 (−404), 258 (−2.03), 277 (−68.5) |
| 5CLB | 284 (2.31 ± 1.48), 204 (6.99 ± 1.33) | 208 (−233), 232 (−7.77), 260 (+70), 286 (+2.42) |
| 56CLB | 291 (2.53 ± 1.23), 204 (8.25 ± 1.77) | 207 (−259), 234 (−138), 240 (−375), 260 (+581), 291 (−224) |
| Platinum(IV)-CLB Complexes | Solvent Systems, Time Points, and HPLC Peak Areas at 254 nm | |||||
|---|---|---|---|---|---|---|
| PBS (~7.4 pH) | 0.9% NaCl | d.i.H2O/DMSO | ||||
| 30 min | 1 h | 30 min | 1 h | 30 min | 1 h | |
| PCLB | 85.0% | 83.1% | 89.4% | 87.5% | 93.2% | 92.4% |
| 5CLB | 68.8% | 67.3% | 73.9% | 72.4% | 76.5% | 75.8% |
| 56CLB | 78.2% | 75.2% | 84.6% | 82.5% | 85.0% | 84.3% |
| Compounds | log kw |
|---|---|
| PCLB | 1.80 |
| 5CLB | 1.87 |
| 56CLB | 1.90 |
| CLB | 1.88 |
| GI50 Values (nM) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Platinum(IV)-CLB Prodrugs | HT29 | U87 | MCF-7 | A2780 | H460 | A431 | Du145 | BE2-C | SJ-G2 | MIA | MCF10A | ADDP | Average GI50 Values |
| PCLB | 170 ± 0.046 | 1200 ± 0.28 | 680 ± 0.043 | 300 ± 0.048 | 460 ± 0.063 | 510 ± 0.10 | 210 ± 0.12 | 440 ± 0.030 | 320 ± 0.040 | 190 ± 0.032 | 300 ± 0.045 | 290 ± 0.037 | 423 ± 0.07 |
| 5CLB | 19 ± 0.0054 | 250 ± 0.015 | 180 ± 0.017 | 46 ± 0.015 | 39 ± 0.013 | 90 ± 0.015 | 14 ± 0.004 | 230 ± 0.039 | 180 ± 0.02 | 34 ± 0.005 | 22 ± 0.0045 | 26 ± 0.0064 | 94 ± 0.01 |
| 56CLB | 6.1 ± 0.0020 | 93 ± 0.0067 | 63 ± 0.015 | 21 ± 0.0047 | 13 ± 0.0056 | 34 ± 0.0037 | 2.7 ± 0.0012 | 140 ± 0.012 | 130 ± 0.03 | 11 ± 0.003 | 7 ± 0.004 | 10 ± 0.0028 | 44 ± 0.01 |
| Platinum(II) complexes | |||||||||||||
| PHENSS | 160 ± 0.045 | 980 ± 0.27 | 1500 ± 0.50 | 230 ± 0.030 | 360 ± 0.035 | 480 ± 0.17 | 100 ± 0.038 | 380 ± 0.046 | 330 ± 0.066 | 200 ± 0.057 | 300 ± 0.058 | 190 ± 0.047 | 434 ± 0.11 |
| 5MESS | 33 ± 0.0038 | 320 ± 0.026 | 200 ± 0.012 | 61 ± 0.010 | 41 ± 0.005 | 120 ± 0.025 | 22 ± 0.0027 | 270 ± 0.038 | 220 ± 0.01 | 48 ± 0.002 | 30 ± 0.0018 | 34 ± 0.0023 | 117 ± 0.01 |
| 56MESS | 10 ± 0.0016 | 35 ± 0.0064 | 93 ± 0.044 | 76 ± 0.057 | 21 ± 0.0019 | 29 ± 0.0010 | 4.6 ± 0.00039 | 59 ± 0.0041 | 66 ± 0.022 | 13 ± 0.0020 | 16 ± 0.0012 | 13 ± 0.0022 | 36 ± 0.01 |
| Platinum(IV) dihydroxy complexes | |||||||||||||
| PHENSS(IV)(OH)2 | 710 ± 0.30 | 4900 ± 0.61 | 16,000 ± 4.5 | 800 ± 0.084 | 1700 ± 0.20 | 4300 ± 0.53 | 310 ± 0.092 | 3000 ± 0.53 | 1700 ± 0.35 | 3400 ± 2.2 | 1700 ± 0.20 | 1300 ± 0.35 | 3318 ± 0.88 |
| 5MESS(IV)(OH)2 | 60 ± 0.0055 | 900 ± 0.058 | 1200 ± 0.39 | 240 ± 0.009 | 60 ± 0.005 | 360 ± 0.058 | 41 ± 0.0049 | 1400 ± 0.3 | 640 ± 0.07 | 160 ± 0.029 | 130 ± 0.019 | 130 ± 0.022 | 443 ± 0.08 |
| 56MESS(IV)(OH)2 | 36 ± 0.0071 | 190 ± 0.023 | 480 ± 0.14 | 59 ± 0.0071 | 190 ± 0.15 | 120 ± 0.022 | 15 ± 0.0026 | 240 ± 0.022 | 210 ± 0.045 | 43 ± 0.0025 | 61 ± 0.0073 | 170 ± 0.12 | 151 ± 0.05 |
| DNA-targeting chemotherapeutics | |||||||||||||
| Cisplatin | 11,300 ± 1.9 | 3800 ± 1.1 | 6500 ± 0.8 | 1000 ± 0.1 | 900 ± 0.2 | 2400 ± 0.3 | 1200 ± 0.1 | 1900 ± 0.2 | 400 ± 0.1 | 7500 ± 1.3 | 5200 ± 0.52 | 28,000 ± 1.6 | 5842 ± 0.61 |
| Oxaliplatin | 900 ± 0.2 | 1800 ± 0.2 | 500 ± 0.1 | 160 ± 0.1 | 1600 ± 0.1 | 4100 ± 0.5 | 2900 ± 0.4 | 900 ± 0.2 | 3000 ± 1.2 | 900 ± 0.2 | nd | 800 ± 0.1 | 1463 ± 0.32 |
| Carboplatin | >50,000 | >50,000 | >50,000 | 9200 ± 2.9 | 14,000 ± 1.0 | 24,000 ± 2.2 | 15,000 ± 1.2 | 19,000 ± 1.2 | 5700 ± 0.2 | >50,000 | >50,000 | >50,000 | 32,242 ± 1.45 |
| CLB | 39,000 ± 2 | 38,000 ± 2 | 11,000 ± 1.9 | 4300 ± 0.4 | 11,000 ± 2 | 35,000 ± 0 | 27,000 ± 3 | 20,000 ± 1 | 11,000 ± 2 | 40,000 ± 2 | 17,000 ± 1 | 16,000 ± 1 | 22,000 ± 1.52 |
| Compounds | ROS Production in Different Time Intervals (RFU) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Control | 50 ± 2 | 52 ± 4 | 57 ± 5 |
| PHENSS | 174 ± 2 | 172 ± 9 | 176 ± 7 |
| 5MESS | 204 ± 4 | 205 ± 3 | 188 ± 3 |
| 56MESS | 240 ± 5 | 218 ± 3 | 255 ± 4 |
| PHENSS(IV)(OH)2 | 144 ± 5 | 273 ± 4 | 303 ± 1 |
| 5MESS(IV)(OH)2 | 234 ± 1 | 323 ± 9 | 335 ± 2 |
| 56MESS(IV)(OH)2 | 259 ± 3 | 356 ± 11 | 438 ± 7 |
| CLB | 110 ± 3 | 190 ± 5 | 204 ± 4 |
| PCLB | 190 ± 2 | 244 ± 7 | 285 ± 3 |
| 5CLB | 337 ± 8 | 324 ± 5 | 376 ± 1 |
| 56CLB | 395 ± 3 | 437 ± 2 | 558 ± 2 |
| Cisplatin | 214 ± 9 | 214 ± 9 | 200 ± 9 |
| TBHP | 514 ± 3 | 336 ± 2 | 332 ± 5 |
| NAC | 71 ± 2 | 85 ± 6 | 91 ± 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aputen, A.D.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Potent Chlorambucil-Platinum(IV) Prodrugs. Int. J. Mol. Sci. 2022, 23, 10471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810471
Aputen AD, Elias MG, Gilbert J, Sakoff JA, Gordon CP, Scott KF, Aldrich-Wright JR. Potent Chlorambucil-Platinum(IV) Prodrugs. International Journal of Molecular Sciences. 2022; 23(18):10471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810471
Chicago/Turabian StyleAputen, Angelico D., Maria George Elias, Jayne Gilbert, Jennette A. Sakoff, Christopher P. Gordon, Kieran F. Scott, and Janice R. Aldrich-Wright. 2022. "Potent Chlorambucil-Platinum(IV) Prodrugs" International Journal of Molecular Sciences 23, no. 18: 10471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810471