
Extended N-Terminal Acetyltransferase Naa50 in Filamentous Fungi Adds to Naa50 Diversity
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Chaetomium thermophilum and Neurospora crassa Naa50 Have Elongated Termini
2.2. Deletion of naa50 or NatA Genes Leads to Neurospora crassa Growth Defects
2.3. Naa50 Acetylates Canonical NatC/E/F-Type Substrates
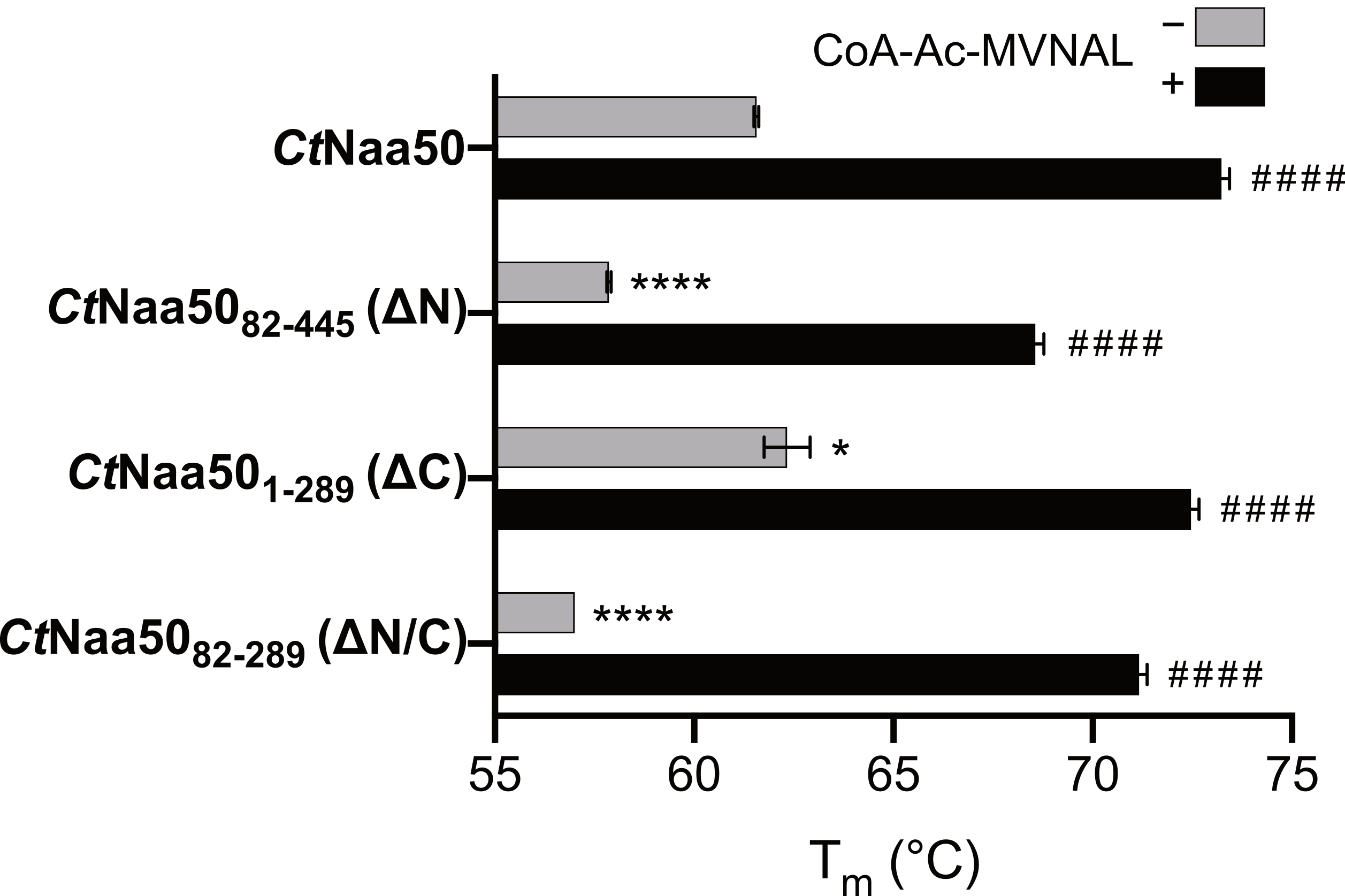
2.4. The N-Terminus of CtNaa50 Stabilizes the GNAT Domain
2.5. Crystal Structure of CtNaa50 with a Bisubstrate Analog
2.6. C. thermophilum Naa50 Does Not Bind to NatA
2.7. C. thermophilum Naa50 Binds to Dynein Light Chain 1 Protein
2.8. Naa50 Binds to the Ribosome with Its C-Terminus
3. Discussion
4. Materials and Methods
4.1. Neurospora crassa Race Tube Growth Phenotypes
4.2. Plasmid Construction
4.3. Expression in E. coli and Purification of Proteins
4.4. Expression in C. thermophilum and Purification of Proteins
4.5. Expression in S. cerevisiae and Purification of Proteins
4.6. Yeast Two-Hybrid (Y2H) Assays
4.7. Analytical Size-Exclusion Chromatography
4.8. Crystallization of CtNaa5082-289/CoA-Ac-MVNAL
4.9. Data Collection, Structure Determination, and Analyses
4.10. Colorimetric In Vitro Activity Assays
4.11. Nano Differential Scanning Fluorimetry (nanoDSF)
4.12. Statistical Analyses of In Vitro Acetylation and Melting Temperatures
4.13. LC-MS/MS Analysis
4.14. C. thermophilum 80S Ribosome Preparation
4.15. Flag-Tag Pull-Down Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varland, S.; Osberg, C.; Arnesen, T. N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics 2015, 15, 2385–2401. [Google Scholar] [CrossRef] [PubMed]
- Aksnes, H.; Drazic, A.; Marie, M.; Arnesen, T. First Things First: Vital Protein Marks by N-Terminal Acetyltransferases. Trends Biochem. Sci. 2016, 41, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics Analyses Reveal the Evolutionary Conservation and Divergence of N-Terminal Acetyltransferases from Yeast and Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef]
- Bienvenut, W.V.; Sumpton, D.; Martinez, A.; Lilla, S.; Espagne, C.; Meinnel, T.; Giglione, C. Comparative Large Scale Characterization of Plant versus Mammal Proteins Reveals Similar and Idiosyncratic N-alpha-Acetylation Features. Mol. Cell. Proteom. 2012, 11, M111.015131. [Google Scholar] [CrossRef] [PubMed]
- Lange, P.F.; Huesgen, P.F.; Nguyen, K.; Overall, C.M. Annotating N termini for the human proteome project: N termini and Nalpha-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome. J. Proteome Res. 2014, 13, 2028–2044. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Marmorstein, R. Protein N-Terminal Acetylation: Structural Basis, Mechanism, Versatility, and Regulation. Trends Biochem. Sci. 2021, 46, 15–27. [Google Scholar] [CrossRef]
- Aksnes, H.; Ree, R.; Arnesen, T. Co-translational, Post-translational, and Non-catalytic Roles of N-Terminal Acetyltransferases. Mol. Cell 2019, 73, 1097–1114. [Google Scholar] [CrossRef]
- Dikiy, I.; Eliezer, D. N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound alpha-synuclein and increases its affinity for physiological membranes. J. Biol. Chem. 2014, 289, 3652–3665. [Google Scholar] [CrossRef]
- Forte, G.M.A.; Pool, M.R.; Stirling, C.J. N-Terminal Acetylation Inhibits Protein Targeting to the Endoplasmic Reticulum. PLoS Biol. 2011, 9, e1001073. [Google Scholar] [CrossRef]
- Holmes, W.M.; Mannakee, B.K.; Gutenkunst, R.N.; Serio, T.R. Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nat. Commun. 2014, 5, 4383. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.S.; Shemorry, A.; Varshavsky, A. N-Terminal Acetylation of Cellular Proteins Creates Specific Degradation Signals. Science 2010, 327, 973–977. [Google Scholar] [CrossRef]
- Lee, C.C.; Peng, S.H.; Shen, L.; Lee, C.F.; Du, T.H.; Kang, M.L.; Xu, G.L.; Upadhyay, A.K.; Cheng, X.; Yan, Y.T.; et al. The Role of N-alpha-acetyltransferase 10 Protein in DNA Methylation and Genomic Imprinting. Mol. Cell 2017, 68, 89–103.e7. [Google Scholar] [CrossRef] [PubMed]
- Linster, E.; Wirtz, M. N-terminal acetylation: An essential protein modification emerges as an important regulator of stress responses. J. Exp. Bot. 2018, 69, 4555–4568. [Google Scholar] [CrossRef] [PubMed]
- Kalvik, T.V.; Arnesen, T. Protein N-terminal acetyltransferases in cancer. Oncogene 2013, 32, 269–276. [Google Scholar] [CrossRef]
- Myklebust, L.M.; Van Damme, P.; Stove, S.I.; Dorfel, M.J.; Abboud, A.; Kalvik, T.V.; Grauffel, C.; Jonckheere, V.; Wu, Y.; Swensen, J.; et al. Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum. Mol. Genet. 2015, 24, 1956–1976. [Google Scholar] [CrossRef]
- Rope, A.F.; Wang, K.; Evjenth, R.; Xing, J.; Johnston, J.J.; Swensen, J.J.; Johnson, W.E.; Moore, B.; Huff, C.D.; Bird, L.M.; et al. Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am. J. Hum. Genet. 2011, 89, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Saunier, C.; Stove, S.I.; Popp, B.; Gerard, B.; Blenski, M.; AhMew, N.; de Bie, C.; Goldenberg, P.; Isidor, B.; Keren, B.; et al. Expanding the Phenotype Associated with NAA10-Related N-Terminal Acetylation Deficiency. Hum. Mutat. 2016, 37, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Aksnes, H.; Van Damme, P.; Goris, M.; Starheim, K.K.; Marie, M.; Stove, S.I.; Hoel, C.; Kalvik, T.V.; Hole, K.; Glomnes, N.; et al. An organellar nalpha-acetyltransferase, naa60, acetylates cytosolic N termini of transmembrane proteins and maintains Golgi integrity. Cell Rep. 2015, 10, 1362–1374. [Google Scholar] [CrossRef]
- Aksnes, H.; Goris, M.; Stromland, O.; Drazic, A.; Waheed, Q.; Reuter, N.; Arnesen, T. Molecular Determinants of the N-Terminal Acetyltransferase Naa60 Anchoring to the Golgi Membrane. J. Biol. Chem. 2017, 292, 6821–6837. [Google Scholar] [CrossRef]
- Chen, J.Y.; Liu, L.; Cao, C.L.; Li, M.J.; Tan, K.; Yang, X.; Yun, C.H. Structure and function of human Naa60 (NatF), a Golgi-localized bi-functional acetyltransferase. Sci. Rep. 2016, 6, 31425. [Google Scholar] [CrossRef] [Green Version]
- Linster, E.; Layer, D.; Bienvenut, W.V.; Dinh, T.V.; Weyer, F.A.; Leemhuis, W.; Brunje, A.; Hoffrichter, M.; Miklankova, P.; Kopp, J.; et al. The Arabidopsis N(alpha) -acetyltransferase NAA60 locates to the plasma membrane and is vital for the high salt stress response. New Phytol. 2020, 228, 554–569. [Google Scholar] [CrossRef] [PubMed]
- Stove, S.I.; Magin, R.S.; Foyn, H.; Haug, B.E.; Marmorstein, R.; Arnesen, T. Crystal Structure of the Golgi-Associated Human Nalpha-Acetyltransferase 60 Reveals the Molecular Determinants for Substrate-Specific Acetylation. Structure 2016, 24, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.V.; Bienvenut, W.V.; Linster, E.; Feldman-Salit, A.; Jung, V.A.; Meinnel, T.; Hell, R.; Giglione, C.; Wirtz, M. Molecular identification and functional characterization of the first N-acetyltransferase in plastids by global acetylome profiling. Proteomics 2015, 15, 2426–2435. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Aksnes, H.; Marie, M.; Boczkowska, M.; Varland, S.; Timmerman, E.; Foyn, H.; Glomnes, N.; Rebowski, G.; Impens, F.; et al. NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc. Natl. Acad. Sci. USA 2018, 115, 4399–4404. [Google Scholar] [CrossRef]
- Goris, M.; Magin, R.S.; Foyn, H.; Myklebust, L.M.; Varland, S.; Ree, R.; Drazic, A.; Bhambra, P.; Stove, S.I.; Baumann, M.; et al. Structural determinants and cellular environment define processed actin as the sole substrate of the N-terminal acetyltransferase NAA80. Proc. Natl. Acad. Sci. USA 2018, 115, 4405–4410. [Google Scholar] [CrossRef]
- Arnesen, T.; Marmorstein, R.; Dominguez, R. Actin’s N-terminal acetyltransferase uncovered. Cytoskeleton 2018, 75, 318–322. [Google Scholar] [CrossRef]
- Rebowski, G.; Boczkowska, M.; Drazic, A.; Ree, R.; Goris, M.; Arnesen, T.; Dominguez, R. Mechanism of actin N-terminal acetylation. Sci. Adv. 2020, 6, eaay8793. [Google Scholar] [CrossRef]
- Arnesen, T.; Starheim, K.K.; Van Damme, P.; Evjenth, R.; Dinh, H.; Betts, M.J.; Ryningen, A.; Vandekerckhove, J.; Gevaert, K.; Anderson, D. The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Mol. Cell. Biol. 2010, 30, 1898–1909. [Google Scholar] [CrossRef]
- Weyer, F.A.; Gumiero, A.; Lapouge, K.; Bange, G.; Kopp, J.; Sinning, I. Structural basis of HypK regulating N-terminal acetylation by the NatA complex. Nat. Commun. 2017, 8, 15726. [Google Scholar] [CrossRef]
- Polevoda, B.; Sherman, F. NatC Nalpha-terminal acetyltransferase of yeast contains three subunits, Mak3p, Mak10p, and Mak31p. J. Biol. Chem. 2001, 276, 20154–20159. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, S.; Hopf, L.V.M.; Bock-Bierbaum, T.; Lally, C.C.M.; Spahn, C.M.T.; Daumke, O. Divergent architecture of the heterotrimeric NatC complex explains N-terminal acetylation of cognate substrates. Nat. Commun. 2020, 11, 5506. [Google Scholar] [CrossRef] [PubMed]
- Polevoda, B.; Norbeck, J.; Takakura, H.; Blomberg, A.; Sherman, F. Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 1999, 18, 6155–6168. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Lasa, M.; Polevoda, B.; Gazquez, C.; Elosegui-Artola, A.; Kim, D.S.; De Juan-Pardo, E.; Demeyer, K.; Hole, K.; Larrea, E.; et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc. Natl. Acad. Sci. USA 2012, 109, 12449–12454. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, M.; Just, S.; Mun, A.; Ross, S.; Rücknagel, P.; Dubaquié, Y.; Ehrenhofer-Murray, A.; Rospert, S. The yeast N(alpha)-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell. Biol. 2003, 23, 7403–7414. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; LP, S.d.C.; Yu, M.; Hegde, S.S.; Magnet, S.; Roderick, S.L.; Blanchard, J.S. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 2005, 433, 212–226. [Google Scholar] [CrossRef]
- Salah Ud-Din, A.I.; Tikhomirova, A.; Roujeinikova, A. Structure and Functional Diversity of GCN5-Related N-Acetyltransferases (GNAT). Int. J. Mol. Sci. 2016, 17, 1018. [Google Scholar] [CrossRef]
- Abboud, A.; Bedoucha, P.; Byska, J.; Arnesen, T.; Reuter, N. Dynamics-function relationship in the catalytic domains of N-terminal acetyltransferases. Comput. Struct. Biotechnol. J. 2020, 18, 532–547. [Google Scholar] [CrossRef]
- Marmorstein, R.; Trievel, R.C. Histone modifying enzymes: Structures, mechanisms, and specificities. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2009, 1789, 58–68. [Google Scholar] [CrossRef]
- Weidenhausen, J.; Kopp, J.; Armbruster, L.; Wirtz, M.; Lapouge, K.; Sinning, I. Structural and functional characterization of the N-terminal acetyltransferase Naa50. Structure 2021, 29, 413–425. [Google Scholar] [CrossRef]
- Magin, R.S.; March, Z.M.; Marmorstein, R. The N-terminal Acetyltransferase Naa10/ARD1 Does Not Acetylate Lysine Residues. J. Biol. Chem. 2016, 291, 5270–5277. [Google Scholar] [CrossRef] [Green Version]
- Rojas, J.R.; Trievel, R.C.; Zhou, J.; Mo, Y.; Li, X.; Berger, S.L.; Allis, C.D.; Marmorstein, R. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature 1999, 401, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Reddi, R.; Saddanapu, V.; Chinthapalli, D.K.; Sankoju, P.; Sripadi, P.; Addlagatta, A. Human Naa50 Protein Displays Broad Substrate Specificity for Amino-terminal Acetylation: DETAILED STRUCTURAL AND BIOCHEMICAL ANALYSIS USING TETRAPEPTIDE LIBRARY. J. Biol. Chem. 2016, 291, 20530–20538. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Hole, K.; Gevaert, K.; Arnesen, T. N-terminal acetylome analysis reveals the specificity of Naa50 (Nat5) and suggests a kinetic competition between N-terminal acetyltransferases and methionine aminopeptidases. Proteomics 2015, 15, 2436–2446. [Google Scholar] [CrossRef]
- Hou, F.; Chu, C.W.; Kong, X.; Yokomori, K.; Zou, H. The acetyltransferase activity of San stabilizes the mitotic cohesin at the centromeres in a shugoshin-independent manner. J. Cell Biol. 2007, 177, 587–597. [Google Scholar] [CrossRef]
- Deng, S.; Magin, R.S.; Wei, X.; Pan, B.; Petersson, E.J.; Marmorstein, R. Structure and Mechanism of Acetylation by the N-Terminal Dual Enzyme NatA/Naa50 Complex. Structure 2019, 27, 1057–1070.e4. [Google Scholar] [CrossRef]
- Williams, B.C.; Garrett-Engele, C.M.; Li, Z.; Williams, E.V.; Rosenman, E.D.; Goldberg, M.L. Two Putative Acetyltransferases, San and Deco, Are Required for Establishing Sister Chromatid Cohesion in Drosophila. Curr. Biol. 2003, 13, 2025–2036. [Google Scholar] [CrossRef]
- Rong, Z.; Ouyang, Z.; Magin, R.S.; Marmorstein, R.; Yu, H. Opposing Functions of the N-terminal Acetyltransferases Naa50 and NatA in Sister-chromatid Cohesion. J. Biol. Chem. 2016, 291, 19079–19091. [Google Scholar] [CrossRef]
- Armbruster, L.; Linster, E.; Boyer, J.B.; Brunje, A.; Eirich, J.; Stephan, I.; Bienvenut, W.V.; Weidenhausen, J.; Meinnel, T.; Hell, R.; et al. NAA50 Is an Enzymatically Active N (alpha)-Acetyltransferase That Is Crucial for Development and Regulation of Stress Responses. Plant Physiol. 2020, 183, 1502–1516. [Google Scholar] [CrossRef]
- Feng, J.; Hu, J.; Li, Y.; Li, R.; Yu, H.; Ma, L. The N-Terminal Acetyltransferase Naa50 Regulates Arabidopsis Growth and Osmotic Stress Response. Plant Cell Physiol. 2020, 61, 1565–1575. [Google Scholar] [CrossRef]
- Neubauer, M.; Innes, R.W. Loss of the Acetyltransferase NAA50 Induces Endoplasmic Reticulum Stress and Immune Responses and Suppresses Growth. Plant Physiol. 2020, 183, 1838–1854. [Google Scholar] [CrossRef]
- Liszczak, G.; Arnesen, T.; Marmorstein, R. Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J. Biol. Chem. 2011, 286, 37002–37010. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; McTiernan, N.; Wei, X.; Arnesen, T.; Marmorstein, R. Molecular basis for N-terminal acetylation by human NatE and its modulation by HYPK. Nat. Commun. 2020, 11, 818. [Google Scholar] [CrossRef] [PubMed]
- Knorr, A.G.; Schmidt, C.; Tesina, P.; Berninghausen, O.; Becker, T.; Beatrix, B.; Beckmann, R. Ribosome-NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nat. Struct. Mol. Biol. 2019, 26, 35–39. [Google Scholar] [CrossRef]
- Rathore, O.S.; Faustino, A.; Prudencio, P.; Van Damme, P.; Cox, C.J.; Martinho, R.G. Absence of N-terminal acetyltransferase diversification during evolution of eukaryotic organisms. Sci. Rep. 2016, 6, 21304. [Google Scholar] [CrossRef]
- Magin, R.S.; Liszczak, G.P.; Marmorstein, R. The molecular basis for histone H4- and H2A-specific amino-terminal acetylation by NatD. Structure 2015, 23, 332–341. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Roche, C.M.; Loros, J.J.; McCluskey, K.; Glass, N.L. Neurospora crassa: Looking back and looking forward at a model microbe. Am. J. Bot. 2014, 101, 2022–2035. [Google Scholar] [CrossRef]
- Starheim, K.K.; Arnesen, T.; Gromyko, D.; Ryningen, A.; Varhaug, J.E.; Lillehaug, J.R. Identification of the human N(alpha)-acetyltransferase complex B (hNatB): A complex important for cell-cycle progression. Biochem. J. 2008, 415, 325–331. [Google Scholar] [CrossRef]
- Van Damme, P.; Hole, K.; Pimenta-Marques, A.; Helsens, K.; Vandekerckhove, J.; Martinho, R.G.; Gevaert, K.; Arnesen, T. NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 2011, 7, e1002169. [Google Scholar] [CrossRef] [Green Version]
- Foyn, H.; Van Damme, P.; Stove, S.I.; Glomnes, N.; Evjenth, R.; Gevaert, K.; Arnesen, T. Protein N-terminal Acetyltransferases Act as N-terminal Propionyltransferases In Vitro and In Vivo. Mol. Cell. Proteom. 2013, 12, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Anderson, D.; Torsvik, J.; Halseth, H.B.; Varhaug, J.E.; Lillehaug, J.R. Cloning and characterization of hNAT5/hSAN: An evolutionarily conserved component of the NatA protein N-alpha-acetyltransferase complex. Gene 2006, 371, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Reck-Peterson, S.L.; Redwine, W.B.; Vale, R.D.; Carter, A.P. The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 2018, 19, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Miskin, J.; Hernaez, B.; Fernandez-Zapatero, P.; Soto, L.; Canto, C.; Rodriguez-Crespo, I.; Dixon, L.; Escribano, J.M. African swine fever virus protein p54 interacts with the microtubular motor complex through direct binding to light-chain dynein. J. Virol. 2001, 75, 9819–9827. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.W.; Kan, H.M.; Chan, L.N.; Xu, W.G.; Wang, K.P.; Wu, Z.; Sheng, M.; Zhang, M. The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J. Biol. Chem. 2005, 280, 8172–8179. [Google Scholar] [CrossRef]
- Myllykoski, M.; Eichel, M.A.; Jung, R.B.; Kelm, S.; Werner, H.B.; Kursula, P. High-affinity heterotetramer formation between the large myelin-associated glycoprotein and the dynein light chain DYNLL1. J. Neurochem. 2018, 147, 764–783. [Google Scholar] [CrossRef]
- Singh, P.K.; Weber, A.; Hacker, G. The established and the predicted roles of dynein light chain in the regulation of mitochondrial apoptosis. Cell Cycle 2018, 17, 1037–1047. [Google Scholar] [CrossRef]
- Lo, K.W.; Naisbitt, S.; Fan, J.S.; Sheng, M.; Zhang, M. The 8-kDa dynein light chain binds to its targets via a conserved (K/R)XTQT motif. J. Biol. Chem. 2001, 276, 14059–14066. [Google Scholar] [CrossRef]
- van Noort, V.; Bradatsch, B.; Arumugam, M.; Amlacher, S.; Bange, G.; Creevey, C.; Falk, S.; Mende, D.R.; Sinning, I.; Hurt, E.; et al. Consistent mutational paths predict eukaryotic thermostability. BMC Evol. Biol. 2013, 13, 7. [Google Scholar] [CrossRef]
- Linster, E.; Stephan, I.; Bienvenut, W.V.; Maple-Grodem, J.; Myklebust, L.M.; Huber, M.; Reichelt, M.; Sticht, C.; Moller, S.G.; Meinnel, T.; et al. Downregulation of N-terminal acetylation triggers ABA-mediated drought responses in Arabidopsis. Nat. Commun. 2015, 6, 7640. [Google Scholar] [CrossRef] [Green Version]
- Bock, T.; Chen, W.H.; Ori, A.; Malik, N.; Silva-Martin, N.; Huerta-Cepas, J.; Powell, S.T.; Kastritis, P.L.; Smyshlyaev, G.; Vonkova, I.; et al. An integrated approach for genome annotation of the eukaryotic thermophile Chaetomium thermophilum. Nucleic Acids Res. 2014, 42, 13525–13533. [Google Scholar] [CrossRef] [PubMed]
- Barbar, E. Dynein Light Chain LC8 Is a Dimerization Hub Essential in Diverse Protein Networks. Biochemistry 2008, 47, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Sargent, M.L.; Briggs, W.P.; Woodward, D.O. Circadian nature of a rhythm expressed by an invertaseless strain of Neurospora crassa. Plant Physiol. 1966, 41, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Hole, K.; Van Damme, P.; Dalva, M.; Aksnes, H.; Glomnes, N.; Varhaug, J.E.; Lillehaug, J.R.; Gevaert, K.; Arnesen, T. The human N-alpha-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PLoS ONE 2011, 6, e24713. [Google Scholar] [CrossRef] [PubMed]
- Polevoda, B.; Hoskins, J.; Sherman, F. Properties of Nat4, an Nα-Acetyltransferase of Saccharomyces cerevisiae That Modifies N Termini of Histones H2A and H4. Mol. Cell. Biol. 2009, 29, 2913–2924. [Google Scholar] [CrossRef]
- Magin, R.S.; Deng, S.; Zhang, H.; Cooperman, B.; Marmorstein, R. Probing the interaction between NatA and the ribosome for co-translational protein acetylation. PLoS ONE 2017, 12, e0186278. [Google Scholar] [CrossRef]
- Hristou, A.; Gerlach, I.; Stolle, D.S.; Neumann, J.; Bischoff, A.; Dunschede, B.; Nowaczyk, M.M.; Zoschke, R.; Schunemann, D. Ribosome-Associated Chloroplast SRP54 Enables Efficient Cotranslational Membrane Insertion of Key Photosynthetic Proteins. Plant Cell 2019, 31, 2734–2750. [Google Scholar] [CrossRef]
- Wegrzyn, R.D.; Hofmann, D.; Merz, F.; Nikolay, R.; Rauch, T.; Graf, C.; Deuerling, E. A conserved motif is prerequisite for the interaction of NAC with ribosomal protein L23 and nascent chains. J. Biol. Chem. 2006, 281, 2847–2857. [Google Scholar] [CrossRef]
- Gumiero, A.; Conz, C.; Gese, G.V.; Zhang, Y.; Weyer, F.A.; Lapouge, K.; Kappes, J.; von Plehwe, U.; Schermann, G.; Fitzke, E.; et al. Interaction of the cotranslational Hsp70 Ssb with ribosomal proteins and rRNA depends on its lid domain. Nat. Commun. 2016, 7, 13563. [Google Scholar] [CrossRef]
- Colot, H.V.; Park, G.; Turner, G.E.; Ringelberg, C.; Crew, C.M.; Litvinkova, L.; Weiss, R.L.; Borkovich, K.A.; Dunlap, J.C. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. USA 2006, 103, 10352–10357. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.H. Neurospora: Contributions of a Model Organism; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Froehlich, A.C.; Loros, J.J.; Dunlap, J.C. Rhythmic binding of a WHITE COLLAR-containing complex to the frequency promoter is inhibited by FREQUENCY. Proc. Natl. Acad. Sci. USA 2003, 100, 5914–5919. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Thomson, E.; Bassler, J.; Gnadig, M.; Griesel, S.; Hurt, E. The Exosome Is Recruited to RNA Substrates through Specific Adaptor Proteins. Cell 2015, 162, 1029–1038. [Google Scholar] [CrossRef]
- Dobrev, N.; Ahmed, Y.L.; Sivadas, A.; Soni, K.; Fischer, T.; Sinning, I. The zinc-finger protein Red1 orchestrates MTREC submodules and binds the Mtl1 helicase arch domain. Nat. Commun. 2021, 12, 3456. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Amlacher, S.; Sarges, P.; Flemming, D.; van Noort, V.; Kunze, R.; Devos, D.P.; Arumugam, M.; Bork, P.; Hurt, E. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 2011, 146, 277–289. [Google Scholar] [CrossRef]
- Kellner, N.; Schwarz, J.; Sturm, M.; Fernandez-Martinez, J.; Griesel, S.; Zhang, W.; Chait, B.T.; Rout, M.P.; Kuck, U.; Hurt, E. Developing genetic tools to exploit Chaetomium thermophilum for biochemical analyses of eukaryotic macromolecular assemblies. Sci. Rep. 2016, 6, 20937. [Google Scholar] [CrossRef]
- James, P.; Halladay, J.; Craig, E.A. Genomic Libraries and a Host Strain Designed for Highly Efficient Two-Hybrid Selection in Yeast. Genetics 1996, 144, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Potterton, E.; Briggs, P.; Turkenburg, M.; Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol. Crystallogr 2003, 59, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Andrew Skaff, D.; Miziorko, H.M. A visible wavelength spectrophotometric assay suitable for high-throughput screening of 3-hydroxy-3-methylglutaryl-CoA synthase. Anal. Biochem. 2010, 396, 96–102. [Google Scholar] [CrossRef]
- Moggridge, S.; Sorensen, P.H.; Morin, G.B.; Hughes, C.S. Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. J. Proteome Res. 2018, 17, 1730–1740. [Google Scholar] [CrossRef]
- Hughes, C.S.; Foehr, S.; Garfield, D.A.; Furlong, E.E.; Steinmetz, L.M.; Krijgsveld, J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10, 757. [Google Scholar] [CrossRef]
- Werner, T.; Sweetman, G.; Savitski, M.F.; Mathieson, T.; Bantscheff, M.; Savitski, M.M. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem. 2014, 86, 3594–3601. [Google Scholar] [CrossRef]
- Sridharan, S.; Kurzawa, N.; Werner, T.; Gunthner, I.; Helm, D.; Huber, W.; Bantscheff, M.; Savitski, M.M. Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat. Commun. 2019, 10, 1155. [Google Scholar] [CrossRef] [PubMed]
- Franken, H.; Mathieson, T.; Childs, D.; Sweetman, G.M.; Werner, T.; Togel, I.; Doce, C.; Gade, S.; Bantscheff, M.; Drewes, G.; et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567–1593. [Google Scholar] [CrossRef]
- Savitski, M.M.; Wilhelm, M.; Hahne, H.; Kuster, B.; Bantscheff, M. A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteom. 2015, 14, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; von Heydebreck, A.; Sültmann, H.; Poustka, A.; Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002, 18 (Suppl. S1), S96–S104. [Google Scholar] [CrossRef] [PubMed]
- Kisonaite, M.; Wild, K.; Lapouge, K.; Ruppert, T.; Sinning, I. High-resolution structures of a thermophilic eukaryotic 80S ribosome reveal atomistic details of translocation. Nat. Commun. 2022, 13, 476. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Km (µM) | kcat (min−1) | kcat/Km (min−1µM−1) | CtNaa5082-289 Efficiency (%) |
|---|---|---|---|---|
| CtNaa5082-289 (GNAT) | 52.7 ± 8.6 | 36.4 ± 1.8 | 0.69 ± 0.12 | 100 ± 17 |
| CtNaa5082-445 (ΔN) | 41.2 ± 6.1 | 23.9 ± 1.0 | 0.58 ± 0.09 | 84 ± 13 |
| CtNaa501-289 (ΔC) | 31.9 ± 4.4 | 16.3 ± 0.6 | 0.51 ± 0.07 | 74 ± 10 |
| CtNaa5082-289 Y190F | 22.0 ± 4.0 | 5.6 ± 0.3 | 0.26 ± 0.05 | 38 ± 7 |
| CtNaa5082-289 H235A | 15.3 ± 3.1 | 1.4 ± 0.1 | 0.09 ± 0.02 | 13 ± 3 |
| NcNaa5093-287 (GNAT) | 146.9 ± 12.1 | 95.9 ± 3.0 | 0.65 ± 0.06 | - |
| AtNaa50* | 201.9 ± 30.3 | 15.6 ± 1.2 | 0.08 ± 0.02 | - |
| HsNaa50* | 4.62 ± 0.85 | 5.22 ± 0.23 | 1.13 ± 0.21 | - |
| CtNaa5082-289/CoA-Ac-MVNAL | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Resolution (Å) | 41.67–1.1 (1.14–1.1) |
| Unique reflection | 92854 (9100) |
| a, b, c (Å) | 36.94, 43.65, 140.07 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Rmerge | 0.066 (1.366) |
| Rpim | 0.019 (0.396) |
| Mean (I/σ(I)) | 18.01 (1.45) |
| Multiplicity | 12.7 (12.5) |
| Completeness (%) | 99.88 (99.66) |
| CC1/2 | 1 (0.631) |
| Refinement | |
| Rwork (%) | 14.42 (23.66) |
| Rfree (%) | 16.38 (25.37) |
| RMSD Bond length (Å) | 0.008 |
| RMSD Bond angle (°) | 1.39 |
| Ramachandran favored (%) | 98.14 |
| Ramachandran allowed (%) | 1.86 |
| Ramachandran outliers (%) | 0.0 |
| Rotamer outliers (%) | 0.53 |
| Clashscore | 1.60 |
| Average B factor (Å2) | 20.45 |
| Protein | 18.29 |
| Ligands | 30.29 |
| Solvent | 33.11 |
| PDB identifier | 7OJU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weidenhausen, J.; Kopp, J.; Ruger-Herreros, C.; Stein, F.; Haberkant, P.; Lapouge, K.; Sinning, I. Extended N-Terminal Acetyltransferase Naa50 in Filamentous Fungi Adds to Naa50 Diversity. Int. J. Mol. Sci. 2022, 23, 10805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810805
Weidenhausen J, Kopp J, Ruger-Herreros C, Stein F, Haberkant P, Lapouge K, Sinning I. Extended N-Terminal Acetyltransferase Naa50 in Filamentous Fungi Adds to Naa50 Diversity. International Journal of Molecular Sciences. 2022; 23(18):10805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810805
Chicago/Turabian StyleWeidenhausen, Jonas, Jürgen Kopp, Carmen Ruger-Herreros, Frank Stein, Per Haberkant, Karine Lapouge, and Irmgard Sinning. 2022. "Extended N-Terminal Acetyltransferase Naa50 in Filamentous Fungi Adds to Naa50 Diversity" International Journal of Molecular Sciences 23, no. 18: 10805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810805