
Sequential Targeting of PLK1 and PARP1 Reverses the Resistance to PARP Inhibitors and Enhances Platin-Based Chemotherapy in BRCA-Deficient High-Grade Serous Ovarian Cancer with KRAS Amplification
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. KRAS Gene Expression and Survival of Ovarian Cancer Patients
2.2. Evaluation of the Cytotoxic Effects of Olaparib and BI6727 on BRCA2-Deficient HGSOC Cells with and without KRAS Amplification
2.3. Sequential Inhibition of PLK1 and PARP Sensitizes HGSOC Cells to Olaparib
2.4. PLK1 and PAPR1 Inhibitions Lead to the Persistence of DSBs and Reduce the Survival of BRCA2-Defective HGSOC Cell Lines over the Long Term
2.5. Combined PARP and PLK1 Inhibition Mediates Sensitivity to CARBOPLATIN-Based Chemotherapy
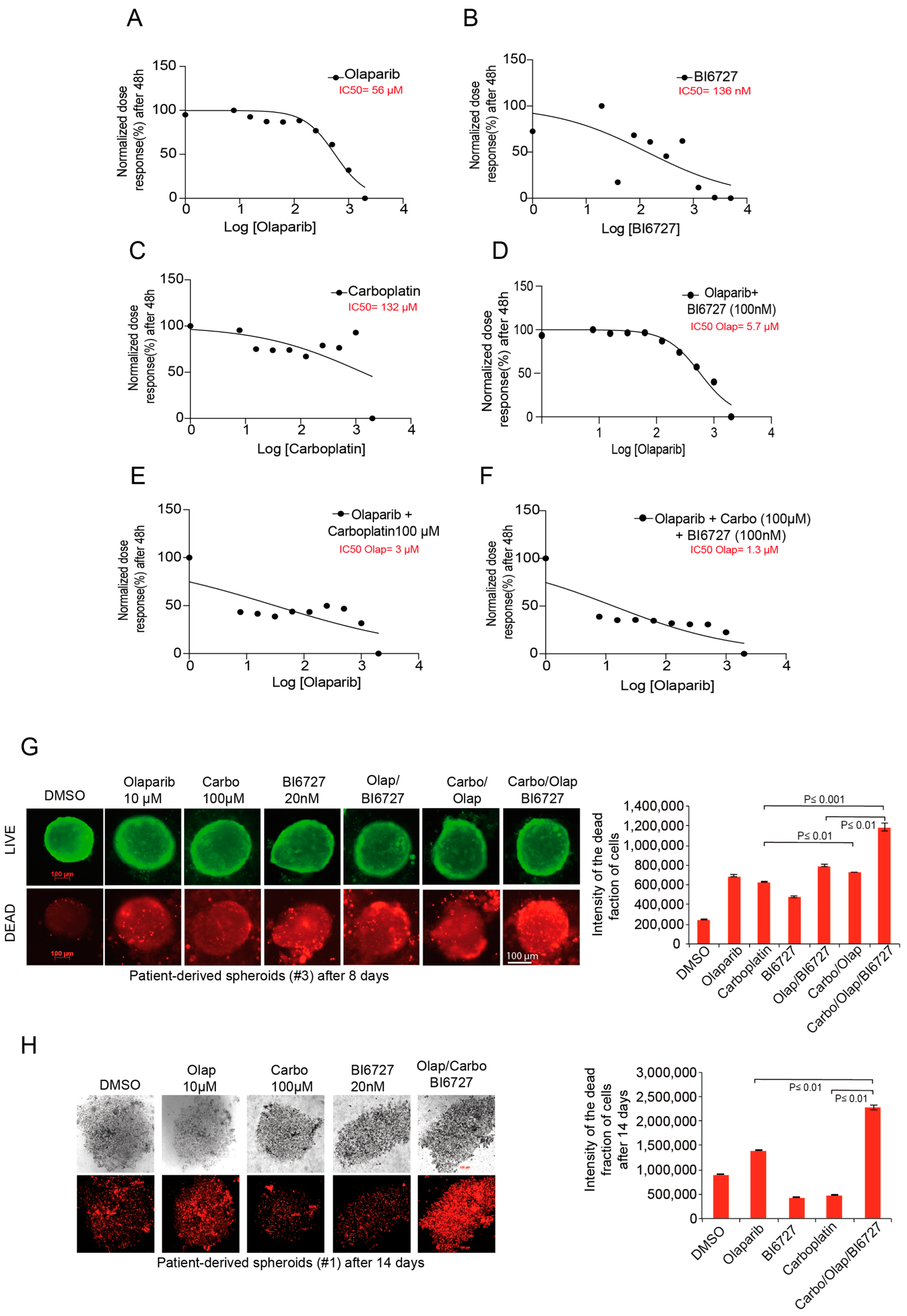
2.6. Inhibition of PLK1 and PARP Increases Carboplatin Response in Ovarian Patient-Derived Spheroid Cultures
3. Discussion
- (1)
- It was shown that resistance to PARP inhibition in HR-deficient HGSOC with amplified KRAS gene and overactivated RAS-RAF-MEK-ERK pathway could be counteracted by sequential inhibition of PARP and PLK1 kinase.
- (2)
- We showed that the resistance of HR-deficient HGSOC cells with KRAS amplification to standard Platinum-based therapy can be reversed by combining sequential PLK1i and PARPi with Carboplatin.
- (3)
- Finally, KRAS amplification could serve as a predictive marker for the efficacy of PLK1i-based combination therapy in HR-deficient ovarian cancer patients.
4. Material and Methods
4.1. Cell Culture
4.2. Patients and Samples (Primary Cell Culture)
4.3. Colony Formation Assay
4.4. Three-Dimensional (3D) Cultures
4.5. Cell Cycle Analysis and Apoptosis Assay
4.6. Genotoxic Treatment
4.7. Cell Cycle-Specific Analysis of DSB Repair
4.8. Immunofluorescence Assays
4.9. Western Blot Analysis
4.10. Antibodies for Western Blot and Chemicals
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Coburn, S.B.; Bray, F.; Sherman, M.E.; Trabert, B. International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Int. J. Cancer 2017, 140, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, W.J.; McGuire, W.P.; Brady, M.F.; Homesley, H.D.; Creasman, W.T.; Berman, M.; Ball, H.; Berek, J.S. The effect of diameter of largest residual disease on survival after primary cytoreductive surgery in patients with suboptimal residual epithelial ovarian carcinoma. Am. J. Obstet. Gynecol. 1994, 170, 974–979; discussion 979–980. [Google Scholar] [CrossRef]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R.; et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef]
- Rahman, M.T.; Nakayama, K.; Rahman, M.; Katagiri, H.; Katagiri, A.; Ishibashi, T.; Ishikawa, M.; Sato, E.; Iida, K.; Nakayama, N.; et al. KRAS and MAPK1 gene amplification in type II ovarian carcinomas. Int. J. Mol. Sci. 2013, 14, 13748–13762. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih Ie, M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef]
- Diaz-Padilla, I.; Malpica, A.L.; Minig, L.; Chiva, L.M.; Gershenson, D.M.; Gonzalez-Martin, A. Ovarian low-grade serous carcinoma: A comprehensive update. Gynecol. Oncol. 2012, 126, 279–285. [Google Scholar] [CrossRef]
- Patton, S.E.; Martin, M.L.; Nelsen, L.L.; Fang, X.; Mills, G.B.; Bast, R.C., Jr.; Ostrowski, M.C. Activation of the ras-mitogen-activated protein kinase pathway and phosphorylation of ets-2 at position threonine 72 in human ovarian cancer cell lines. Cancer Res. 1998, 58, 2253–2259. [Google Scholar]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Walsh, T.; Lee, M.K.; Casadei, S.; Thornton, A.M.; Stray, S.M.; Pennil, C.; Nord, A.S.; Mandell, J.B.; Swisher, E.M.; King, M.C. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 12629–12633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, F.G.; Matuo, R.; Soares, D.G.; Escargueil, A.E.; Henriques, J.A.; Larsen, A.K.; Saffi, J. PARPs and the DNA damage response. Carcinogenesis 2012, 33, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Malyuchenko, N.V.; Kotova, E.Y.; Kulaeva, O.I.; Kirpichnikov, M.P.; Studitskiy, V.M. PARP1 Inhibitors: Antitumor drug design. Acta Nat. 2015, 7, 27–37. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Sisay, M.; Edessa, D. PARP inhibitors as potential therapeutic agents for various cancers: Focus on niraparib and its first global approval for maintenance therapy of gynecologic cancers. Gynecol. Oncol. Res. Pract. 2017, 4, 18. [Google Scholar] [CrossRef]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef]
- Raab, M.; Matthess, Y.; Raab, C.A.; Gutfreund, N.; Dotsch, V.; Becker, S.; Sanhaji, M.; Strebhardt, K. A dimerization-dependent mechanism regulates enzymatic activation and nuclear entry of PLK1. Oncogene 2022, 41, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Sanhaji, M.; Ritter, A.; Belsham, H.R.; Friel, C.T.; Roth, S.; Louwen, F.; Yuan, J. Polo-like kinase 1 regulates the stability of the mitotic centromere-associated kinesin in mitosis. Oncotarget 2014, 5, 3130–3144. [Google Scholar] [CrossRef]
- van de Weerdt, B.C.; Medema, R.H. Polo-like kinases: A team in control of the division. Cell Cycle 2006, 5, 853–864. [Google Scholar] [CrossRef]
- Eckerdt, F.; Yuan, J.; Strebhardt, K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276. [Google Scholar] [CrossRef]
- Mandal, R.; Strebhardt, K. Plk1: Unexpected roles in DNA replication. Cell Res. 2013, 23, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Yata, K.; Lloyd, J.; Maslen, S.; Bleuyard, J.Y.; Skehel, M.; Smerdon, S.J.; Esashi, F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 2012, 45, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, J.; Kong, Y.; Yan, S.; Ahmad, N.; Liu, X. Plk1 Phosphorylation of Mre11 Antagonizes the DNA Damage Response. Cancer Res. 2017, 77, 3169–3180. [Google Scholar] [CrossRef]
- Smits, V.A.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2000, 2, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 2004, 117, 575–588. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Chiappa, M.; Petrella, S.; Damia, G.; Broggini, M.; Guffanti, F.; Ricci, F. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 2022, 12, 903016. [Google Scholar] [CrossRef] [PubMed]
- Rodel, F.; Zhou, S.; Gyorffy, B.; Raab, M.; Sanhaji, M.; Mandal, R.; Martin, D.; Becker, S.; Strebhardt, K. The Prognostic Relevance of the Proliferation Markers Ki-67 and Plk1 in Early-Stage Ovarian Cancer Patients With Serous, Low-Grade Carcinoma Based on mRNA and Protein Expression. Front. Oncol. 2020, 10, 558932. [Google Scholar] [CrossRef]
- Weichert, W.; Denkert, C.; Schmidt, M.; Gekeler, V.; Wolf, G.; Kobel, M.; Dietel, M.; Hauptmann, S. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br. J. Cancer 2004, 90, 815–821. [Google Scholar] [CrossRef]
- Zhang, R.; Shi, H.; Ren, F.; Liu, H.; Zhang, M.; Deng, Y.; Li, X. Misregulation of polo-like protein kinase 1, P53 and P21WAF1 in epithelial ovarian cancer suggests poor prognosis. Oncol. Rep. 2015, 33, 1235–1242. [Google Scholar] [CrossRef]
- Jimeno, A.; Rubio-Viqueira, B.; Rajeshkumar, N.V.; Chan, A.; Solomon, A.; Hidalgo, M. A fine-needle aspirate-based vulnerability assay identifies polo-like kinase 1 as a mediator of gemcitabine resistance in pancreatic cancer. Mol. Cancer Ther. 2010, 9, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.; Bhui, K.; Singh, R.; Singh, M.; Raisuddin, S.; Shukla, Y. Polo-like kinase1 (Plk1) knockdown enhances cisplatin chemosensitivity via up-regulation of p73alpha in p53 mutant human epidermoid squamous carcinoma cells. Biochem. Pharmacol. 2010, 80, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, K.V.; Ferenc, V.; Peter, B.; Gruze, A.; Meyer, R.A.; Hadzijusufovic, E.; Cerny-Reiterer, S.; Mayerhofer, M.; Pickl, W.F.; Sillaber, C.; et al. Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: Overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res. 2010, 70, 1513–1523. [Google Scholar] [CrossRef]
- Noack, S.; Raab, M.; Matthess, Y.; Sanhaji, M.; Kramer, A.; Gyorffy, B.; Kaderali, L.; El-Balat, A.; Becker, S.; Strebhardt, K. Synthetic lethality in CCNE1-amplified high grade serous ovarian cancer through combined inhibition of Polo-like kinase 1 and microtubule dynamics. Oncotarget 2018, 9, 25842–25859. [Google Scholar] [CrossRef]
- Xie, F.F.; Pan, S.S.; Ou, R.Y.; Zheng, Z.Z.; Huang, X.X.; Jian, M.T.; Qiu, J.G.; Zhang, W.J.; Jiang, Q.W.; Yang, Y.; et al. Volasertib suppresses tumor growth and potentiates the activity of cisplatin in cervical cancer. Am. J. Cancer Res. 2015, 5, 3548–3559. [Google Scholar]
- Gyorffy, B.; Lanczky, A.; Szallasi, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr.-Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Prat, J.; FIGO Committee on Gynecologic Oncology. FIGO’s staging classification for cancer of the ovary, fallopian tube, and peritoneum: Abridged republication. J. Gynecol. Oncol. 2015, 26, 87–89. [Google Scholar] [CrossRef]
- Lee, J.M.; Hays, J.L.; Annunziata, C.M.; Noonan, A.M.; Minasian, L.; Zujewski, J.A.; Yu, M.; Gordon, N.; Ji, J.; Sissung, T.M.; et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J. Natl. Cancer Inst. 2014, 106, dju089. [Google Scholar] [CrossRef]
- Meehan, R.S.; Chen, A.P. New treatment option for ovarian cancer: PARP inhibitors. Gynecol. Oncol. Res. Pract. 2016, 3, 3. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e857. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargbo, R.B. Modulation of KRAS Mutant by Inhibiting PLK1 Kinase in Cancer Therapeutics. ACS Med. Chem. Lett. 2021, 12, 1514–1516. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, K.; Guo, J.; Cheng, F.; Lv, J.; Jiang, W.; Lu, W.; Liu, J.; Pang, X.; Liu, M. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat. Commun. 2016, 7, 11363. [Google Scholar] [CrossRef]
- Coulson-Gilmer, C.; Morgan, R.D.; Nelson, L.; Barnes, B.M.; Tighe, A.; Wardenaar, R.; Spierings, D.C.J.; Schlecht, H.; Burghel, G.J.; Foijer, F.; et al. Replication catastrophe is responsible for intrinsic PAR glycohydrolase inhibitor-sensitivity in patient-derived ovarian cancer models. J. Exp. Clin. Cancer Res. 2021, 40, 323. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- van Vugt, M.A.; Bras, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef]
- Bruinsma, W.; Raaijmakers, J.A.; Medema, R.H. Switching Polo-like kinase-1 on and off in time and space. Trends Biochem. Sci. 2012, 37, 534–542. [Google Scholar] [CrossRef]
- Raab, M.; Kobayashi, N.F.; Becker, S.; Kurunci-Csacsko, E.; Kramer, A.; Strebhardt, K.; Sanhaji, M. Boosting the apoptotic response of high-grade serous ovarian cancers with CCNE1 amplification to paclitaxel in vitro by targeting APC/C and the pro-survival protein MCL-1. Int. J. Cancer 2020, 146, 1086–1098. [Google Scholar] [CrossRef]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Lbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Spies, J.; Waizenegger, A.; Barton, O.; Surder, M.; Wright, W.D.; Heyer, W.D.; Lobrich, M. Nek1 Regulates Rad54 to Orchestrate Homologous Recombination and Replication Fork Stability. Mol. Cell 2016, 62, 903–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pariset, E.; Bertucci, A.; Petay, M.; Malkani, S.; Lopez Macha, A.; Paulino Lima, I.G.; Gomez Gonzalez, V.; Tin, A.S.; Tang, J.; Plante, I.; et al. DNA Damage Baseline Predicts Resilience to Space Radiation and Radiotherapy. Cell Rep. 2020, 33, 108434. [Google Scholar] [CrossRef] [PubMed]
- Banath, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual gammaH2AX foci as an indication of lethal DNA lesions. BMC Cancer 2010, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Kraus, W.L. PARP Inhibitors for Cancer Therapy. Cell 2017, 169, 183. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Wilcoxen, K.; Rowley, M.; Toniatti, C. Niraparib: A Poly(ADP-ribose) Polymerase (PARP) Inhibitor for the Treatment of Tumors with Defective Homologous Recombination. J. Med. Chem. 2015, 58, 3302–3314. [Google Scholar] [CrossRef] [PubMed]
- Vanderstichele, A.; Loverix, L.; Busschaert, P.; Van Nieuwenhuysen, E.; Han, S.N.; Concin, N.; Callewaert, T.; Olbrecht, S.; Salihi, R.; Berteloot, P.; et al. Randomized CLIO/BGOG-ov10 trial of olaparib monotherapy versus physician’s choice chemotherapy in relapsed ovarian cancer. Gynecol. Oncol. 2022, 165, 14–22. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Cagnol, S.; Rivard, N. Oncogenic KRAS and BRAF activation of the MEK/ERK signaling pathway promotes expression of dual-specificity phosphatase 4 (DUSP4/MKP2) resulting in nuclear ERK1/2 inhibition. Oncogene 2013, 32, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Wang, Y.; Chen, W.; Yang, S. MicroRNA-508 suppresses epithelial-mesenchymal transition, migration, and invasion of ovarian cancer cells through the MAPK1/ERK signaling pathway. J. Cell Biochem. 2018, 119, 7431–7440. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Yang, J.L. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J. Biol. Chem. 2006, 281, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hou, X.; Shao, C.; Li, J.; Cheng, J.X.; Kuang, S.; Ahmad, N.; Ratliff, T.; Liu, X. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res. 2014, 74, 6635–6647. [Google Scholar] [CrossRef]
- Yim, H.; Erikson, R.L. Plk1-targeted therapies in TP53- or RAS-mutated cancer. Mutat. Res. Rev. Mutat. Res. 2014, 761, 31–39. [Google Scholar] [CrossRef]
- Raab, M.; Sanhaji, M.; Zhou, S.; Rodel, F.; El-Balat, A.; Becker, S.; Strebhardt, K. Blocking Mitotic Exit of Ovarian Cancer Cells by Pharmaceutical Inhibition of the Anaphase-Promoting Complex Reduces Chromosomal Instability. Neoplasia 2019, 21, 363–375. [Google Scholar] [CrossRef]
- Chang, P.; Jacobson, M.K.; Mitchison, T.J. Poly(ADP-ribose) is required for spindle assembly and structure. Nature 2004, 432, 645–649. [Google Scholar] [CrossRef]
- Hyun, S.Y.; Hwang, H.I.; Jang, Y.J. Polo-like kinase-1 in DNA damage response. BMB Rep. 2014, 47, 249–255. [Google Scholar] [CrossRef]
- Prasad, C.B.; Prasad, S.B.; Yadav, S.S.; Pandey, L.K.; Singh, S.; Pradhan, S.; Narayan, G. Olaparib modulates DNA repair efficiency, sensitizes cervical cancer cells to cisplatin and exhibits anti-metastatic property. Sci. Rep. 2017, 7, 12876. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Della Pepa, C.; Tonini, G.; Pisano, C.; Di Napoli, M.; Cecere, S.C.; Tambaro, R.; Facchini, G.; Pignata, S. Ovarian cancer standard of care: Are there real alternatives? Chin. J. Cancer 2015, 34, 17–27. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M.; Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics. REporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res. Treat. 2006, 100, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Helmke, C.; Raab, M.; Rodel, F.; Matthess, Y.; Oellerich, T.; Mandal, R.; Sanhaji, M.; Urlaub, H.; Rodel, C.; Becker, S.; et al. Ligand stimulation of CD95 induces activation of Plk3 followed by phosphorylation of caspase-8. Cell Res. 2016, 26, 914–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasimli, K.; Raab, M.; Tahmasbi Rad, M.; Kurunci-Csacsko, E.; Becker, S.; Strebhardt, K.; Sanhaji, M. Sequential Targeting of PLK1 and PARP1 Reverses the Resistance to PARP Inhibitors and Enhances Platin-Based Chemotherapy in BRCA-Deficient High-Grade Serous Ovarian Cancer with KRAS Amplification. Int. J. Mol. Sci. 2022, 23, 10892. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810892
Gasimli K, Raab M, Tahmasbi Rad M, Kurunci-Csacsko E, Becker S, Strebhardt K, Sanhaji M. Sequential Targeting of PLK1 and PARP1 Reverses the Resistance to PARP Inhibitors and Enhances Platin-Based Chemotherapy in BRCA-Deficient High-Grade Serous Ovarian Cancer with KRAS Amplification. International Journal of Molecular Sciences. 2022; 23(18):10892. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810892
Chicago/Turabian StyleGasimli, Khayal, Monika Raab, Morva Tahmasbi Rad, Elisabeth Kurunci-Csacsko, Sven Becker, Klaus Strebhardt, and Mourad Sanhaji. 2022. "Sequential Targeting of PLK1 and PARP1 Reverses the Resistance to PARP Inhibitors and Enhances Platin-Based Chemotherapy in BRCA-Deficient High-Grade Serous Ovarian Cancer with KRAS Amplification" International Journal of Molecular Sciences 23, no. 18: 10892. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810892