
Spatial Genomic Resource Reveals Molecular Insights into Key Bioactive-Metabolite Biosynthesis in Endangered Angelica glauca Edgew
 ,
,  , , ,
, , ,
Abstract
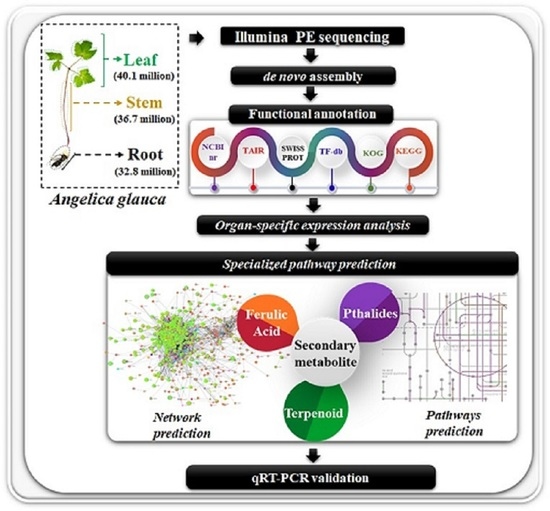
:
1. Introduction
2. Results
2.1. Transcriptome Sequencing, De Novo Assembly, and Functional Annotation
2.2. Differential Transcript Expression and Enrichment Analysis
GO-Enrichment Analysis
2.3. Protein–Protein-Interactome-Network Analysis
2.4. Organ-Specific Gene-Expression Elucidation of Key Metabolite Biosynthesis
2.5. Expression Dynamics of Transcription Factors
2.6. Differential Expressions of CYPs, UGTs, and Transporters
2.7. RNA-Seq Validation Using qRT-PCR
2.8. GC-MS and UPLC Analysis
3. Discussion
3.1. Organ-Specific Transcriptional Dynamics in A. gluaca
3.2. Elucidation of Key Biosynthetic Pathways
4. Methods and Materials
4.1. Plant Materials and RNA Isolation
4.2. Library Construction, Transcriptome Sequencing, and De Novo Assembly
4.3. Functional Annotation of De Novo-Assembled Transcripts
4.4. Differential Expression Analysis
4.5. Protein–Protein-Interactome-Network Prediction
4.6. qRT-PCR Validation of RNA-Seq Data
4.7. Metabolite Profiling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IHR | Indian Himalayan Region |
| MAPs | Medicinal and Aromatic Plant species |
| UGT | UDP-Dependent Glucuronosyltransferase |
| ABC | ATP-binding cassette |
| CYP450 | Cytochrome P450 |
| DXS | 1-deoxy-D-xylulose-5-phosphate synthase |
| DXR | 1-deoxy-D-xylulose 5-phosphate reductoisomerase |
| MCT | 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase |
| CMK | 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase |
| MECS | 2-C-methyl-D-erythritol 2,4-cyclodiphosphatesynthase |
| HDS | 4-hydroxy-3-methylbut-2-enyl diphosphate synthase |
| HDR | 4-hydroxy-3-methylbut-2-enyl diphosphate reductase |
| AACT | Acetyl Co-A acetyltransferase |
| HMGS | HMG-CoA synthase |
| HMGR | HMG-CoA reductase |
| MK | Mevalonate kinase |
| MPK | Phosphomevalonate kinase |
| MVD | Di phosphomevalonate decarboxylase |
| IDI | Isopentenyl-diphosphate delta-isomerase |
| GPPS | Geranyl pyrophosphate synthase |
| FPPS | Fernyl pyrophosphate synthase |
| STS | Sesquiterpene synthase |
| LS | Limonene synthase |
| NMD | Neomenthol dehydrogenase |
| CD | Carveol dehydrogenase |
| NES | Nerolidol synthase |
| DHS | 3-deoxy-D-arabino-hept-2-ulosonate 7-phosphate synthase |
| DHQS | 3-dehydroquinate synthase |
| DHQ-SDH | 3-dehydroquinate dehydratase/shikimate 5-dehydrogenase |
| SK | Shikimate kinase |
| EPSPS | 5-enolpyruvylshikimate-3-phosphate synthase |
| CS | Chorismate synthase |
| CM | Chorismate mutase |
| PAT | Prephenate aminotransferase |
| ADT | Arogenate dehydratase |
| PAL | Phenylalanine ammonia lyase |
| C4H | Cinnamate-4-hydroxylase |
| C3H | Coumarate 3-hydroxylase |
| COMT | Caffeic acid 3-O-methyltransferase |
| NADPH-AOR | NADPH-dependent alkenal/one oxidoreductase |
| SDM | S-adenosyl-L-methionine-dependent Methyltransferases |
| PKC | Polyketide cyclase |
References
- Rodgers, W.A.; Panwar, H.S. Planning a Wildlife Protected Area Network in India; A Report; Wildlife Institute of India: Dehradun, India, 1988.
- Pal, M.; Samant, S.S. Diversity, distribution and conservation of economically important medicinal and aromatic plants of the Indian Himalayan region. Blood Purif. 2005, 1500, 2500. [Google Scholar]
- Kumar, A.; Jnanesha, A.C. Conservation of Rare and Endangered Plant Species for Medicinal. Use. Int. J. Sci. Res. 2016, 5, 1370–1372. [Google Scholar]
- Samarth, R.M.; Samarth, M.; Matsumoto, Y. Medicinally important aromatic plants with radioprotective activity. Future Sci. 2017, 3, 247. [Google Scholar] [CrossRef] [PubMed]
- Gopi, D.K.; Mattummal, R.; Narayana, S.K.K.; Parameswaran, S. IUCN Red Listed Medicinal Plants of Siddha. J. Res. Sid. Med. 2018, 1, 15–22. [Google Scholar]
- Thakur, S.D. Phytochemical constituents of some important medicinal plants from Dachigam National Park, Srinagar, Jammu and Kashmir. Pharma Innov. 2019, 8, 68–71. [Google Scholar]
- Nag, A.; Chanda, S.; Rajkumar, S. Estimation of nuclear genome size of important medicinal plant species from western Himalaya using flow cytometry. J. Cell Plant Sci. 2011, 2, 19–23. [Google Scholar]
- Purohit, V.K.; Andola, H.C.; Haider, S.Z.; Tiwari, D.; Bahuguna, Y.M.; Gairola, K.C.; Arunachalam, K. Essential oil constituents of Angelica glauca Edgew. roots: An endangered species from Uttarakhand Himalaya (India). Natl. Acad. Sci. Lett. 2015, 38, 445–447. [Google Scholar] [CrossRef]
- Joshi, R.K. Angelica (Angelica glauca and A. archangelica) Oils. In Essential Oils in Food Preservation, Flavor and Safety; Academic Press: Cambridge, MA, USA, 2016; pp. 203–208. [Google Scholar] [CrossRef]
- Gautam, K.; Raina, R. Floral architecture, breeding system, seed biology and chromosomal studies in endangered Himalayan Angelica glauca Edgew.(Apiaceae). Caryologia Int. J. Cytol. Cytosystematics Cytogenet. 2019, 72, 23–34. [Google Scholar] [CrossRef]
- Bisht, A.K.; Bhatt, A.; Rawal, R.S.; Dhar, U. Assessment of reproductive potential of different populations of Angelica glauca Edgew., a critically endangered Himalayan medicinal herb. J. Mt. Sci. 2008, 5, 84–90. [Google Scholar] [CrossRef]
- Rawat, J.M.; Bhandari, A.; Mishra, S.; Rawat, B.; Dhakad, A.K.; Thakur, A.; Chandra, A. Genetic stability and phytochemical profiling of the in vitro regenerated plants of Angelica glauca Edgew: An endangered medicinal plant of Himalaya. Plant Cell Tissue Org. Cult. 2018, 135, 111–118. [Google Scholar] [CrossRef]
- Agnihotri, V.K.; Thappa, R.K.; Meena, B.; Kapahi, B.K.; Saxena, R.K.; Qazi, G.N.; Agarwal, S.G. Essential oil composition of aerial parts of Angelica glauca growing wild in North-West Himalaya(India). Phytochemistry 2004, 65, 2411–2413. [Google Scholar] [CrossRef] [PubMed]
- Irshad, M.; Shahid, M.; Aziz, S.; Ghous, T. Antioxidant, antimicrobial and phytotoxic activities of essential oil of Angelica glauca. Asian J. Chem. 2011, 23, 1947. [Google Scholar]
- Chung, J.W.; Choi, R.J.; Seo, E.K.; Nam, J.W.; Dong, M.S.; Shin, E.M.; Guo, L.Y.; Kim, Y.S. Anti-inflammatory effects of (Z)-ligustilide through suppression of mitogen-activated protein kinases and nuclear factor-κB activation pathways. Arch. Pharm. Res. 2012, 35, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Butola, J.S.; Vashistha, R.K. An overview on conservation and utilization of Angelica glauca Edgew. in three Himalayan states of India. Med. Plants-Int. J. Phytomed. Relat. Ind. 2013, 5, 171–178. [Google Scholar] [CrossRef]
- El Maddah, F.; Eguereva, E.; Kehraus, S.; König, G.M. Bios.ynthetic studies of novel polyketides from the marine sponge-derived fungus Stachylidium sp. 293K04Org. Biomol. Chem. 2019, 17, 2747–2752. [Google Scholar] [CrossRef]
- Kong, J.Q. Phenylalanine ammonia-lyase, a key component used for phenylpropanoids production by metabolic engineering. RSC Adv. 2015, 5, 62587–62603. [Google Scholar] [CrossRef]
- Rastogi, S.; Shah, S.; Kumar, R.; Kumar, A.; Shasany, A.K. Comparative temporal metabolomics studies to investigate interspecies variation in three Ocimum species. Sci. Rep. 2020, 10, 5234. [Google Scholar] [CrossRef]
- Henry, L.K.; Thomas, S.T.; Widhalm, J.R.; Lynch, J.H.; Davis, T.C.; Kessler, S.A.; Bohlmann, J.; Noel, J.P.; Dudareva, N. Contribution of isopentenyl phosphate to plant terpenoid metabolism. Nat. Plants 2018, 4, 721–729. [Google Scholar] [CrossRef]
- Sharma, S.; Rasal, V.P.; Patil, P.A.; Joshi, R.K. Effect of Angelica glauca essential oil on allergic airway changes induced by histamine and ovalbumin in experimental animals. Indian J. Pharmacol. 2017, 49, 55. [Google Scholar] [CrossRef]
- Lewin, H.A.; Richards, S.; Aiden, E.L.; Allende, M.L.; Archibald, J.M.; Bálint, M.; Barker, K.B.; Baumgartner, B.; Belov, K.; Bertorelle, G.; et al. The earth BioGenome project 2020: Starting the clock. Proc. Natl. Acad. Sci. USA 2022, 119, e2115635118. [Google Scholar] [CrossRef]
- Unamba, C.I.; Nag, A.; Sharma, R.K. Next generation sequencing technologies: The doorway to the unexplored genomics of non-model plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Wang, X.; Zhong, L.; Tang, Y.; Zhou, X.; Liu, Y.; Zhan, R.; Zheng, H.; Chen, W.; et al. Full-length transcriptome sequencing and methyl jasmonate-induced expression profile analysis of genes related to patchoulol biosynthesis and regulation in Pogostemon cablin. BMC Plant Biol. 2019, 19, 266. [Google Scholar] [CrossRef] [PubMed]
- Pattison, R.J.; Csukasi, F.; Zheng, Y.; Fei, Z.; van der Knaap, E.; Catalá, C. Comprehensive tissue-specific transcriptome analysis reveals distinct regulatory programs during early tomato fruit development. Plant Physiol. 2015, 168, 1684–1701. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Wu, J.; Yi, H. Global tissue-specific transcriptome analysis of Citrus sinensis fruit across six developmental stages. Sci. Data 2019, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Seth, R.; Thakur, S.; Parmar, R.; Masand, M.; Devi, A.; Singh, G.; Dhyani, P.; Choudhary, S.; Sharma, R.K. Genome-wide transcriptional analysis unveils the molecular basis of organ-specific expression of isosteroidal alkaloids biosynthesis in critically endangered Fritillaria roylei Hook. Phytochemistry 2021, 187, 112772. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Q.; Fang, X.; Wu, X.M.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. Transcriptional regulation of plant secondary metabolism. J. Integr. Plant Biol. 2012, 54, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.C.; Kim, W.; Park, S.C.; Jeong, J.; Park, M.K.; Lim, S.; Lee, Y.; Im, W.T.; Lee, J.H.; Choi, G.; et al. Two ginseng UDP-glycosyltransferases synthesize ginsenoside Rg3 and Rd. Plant Cell Physiol. 2014, 55, 2177–2188. [Google Scholar] [CrossRef]
- Naveed, A.; Li, H.; Liu, X. Cytochrome P450s: Blueprints for Potential Applications in Plants. J. Plant Biochem. Physiol. 2018, 6, 204. [Google Scholar] [CrossRef]
- Yazaki, K.; Sugiyama, A.; Morita, M.; Shitan, N. Secondary transport as an efficient membrane transport mechanism for plant secondary metabolites. Phytochem. Rev. 2008, 7, 513–524. [Google Scholar] [CrossRef]
- Younesi-Melerdi, E.; Nematzadeh, G.A.; Pakdin-Parizi, A.; Bakhtiarizadeh, M.R.; Motahari, S.A. De novo RNA sequencing analysis of Aeluropus littoralis halophyte plant under salinity stress. Sci. Res. 2020, 10, 9148. [Google Scholar] [CrossRef]
- Lei, B.; Chang, W.; Zhao, H.; Zhang, K.; Yu, J.; Yu, S.; Cai, K.; Zhang, J.; Lu, K. Nitrogen application and differences in leaf number retained after topping affect the tobacco (Nicotiana tabacum) transcriptome and metabolome. BMC Plant Biol. 2022, 22, 38. [Google Scholar] [CrossRef]
- Pillai, S.G.; Yadav, Y.; Sharma, K.C. A review on an endangered himalayan medicinal aromatic plant-choraka (Angelica glauca edgew). IAMJ 2020, 8, 4509–4515. [Google Scholar] [CrossRef]
- Singh, G.; Chandra, N.; Lingwal, S.; Bisht, M.P.S.; Tiwari, L.M. Distribution and threat assessment of an endemic and endangered species Angelica glauca in high ranges of western Himalaya. J. Herbs Spices Med. Plants 2020, 26, 394–404. [Google Scholar] [CrossRef]
- IUCN. The IUCN Red List of Threatened Species. Version 2021. Available online: http://www.iucnredlist.org (accessed on 7 November 2021).
- Chen, C.; Chen, Y.; Huang, W.; Jiang, Y.; Zhang, H.; Wu, W. Mining of simple sequence repeats (SSRs) loci and development of novel transferability-across EST-SSR markers from de novo transcriptome assembly of Angelica dahurica. PLoS ONE 2019, 14, e0221040. [Google Scholar] [CrossRef]
- Xu, R.; Xu, J.; Li, Y.C.; Dai, Y.T.; Zhang, S.P.; Wang, G.; Liu, Z.G.; Dong, L.L.; Chen, S.L. Integrated chemical and transcriptomic analyses unveil synthetic characteristics of different medicinal root parts of Angelica sinensis. Chin. Herb. Med. 2020, 12, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.P.; Dueholm, B.; Weitzel, C.; Zhang, Y.; Sensen, C.W.; Simonsen, H.T. Transcriptome analysis of Thapsia laciniata Rouy provides insights into terpenoid biosynthesis and diversity in Apiaceae. Int. J. Mol. Sci. 2013, 14, 9080–9098. [Google Scholar] [CrossRef]
- Amini, H.; Naghavi, M.R.; Shen, T.; Wang, Y.; Nasiri, J.; Khan, I.A.; Fiehn, O.; Zerbe, P.; Maloof, J.N. Tissue-specific transcriptome analysis reveals candidate genes for terpenoid and phenylpropanoid metabolism in the medicinal plant Ferula assafoetida. G3 Genes Genomes Genet. 2019, 9, 807–816. [Google Scholar] [CrossRef]
- Lovén, J.; Orlando, D.A.; Sigova, A.A.; Lin, C.Y.; Rahl, P.B.; Burge, C.B.; Levens, D.L.; Lee, T.I.; Young, R.A. Revisiting global gene expression analysis. Cell 2012, 151, 476–482. [Google Scholar] [CrossRef]
- Huang, Q.; Huang, X.; Deng, J.; Liu, H.; Liu, Y.; Yu, K.; Huang, B. Differential gene expression between leaf and rhizome in Atractylodes lancea: A comparative transcriptome analysis. Front. Plant Sci. 2016, 7, 348. [Google Scholar] [CrossRef]
- Wang, T.; Li, B.; Nelson, C.E.; Nabavi, S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinform. 2019, 20, 40. [Google Scholar] [CrossRef]
- Nag, A.; Choudhary, S.; Masand, M.; Parmar, R.; Bhandawat, A.; Seth, R.; Singh, G.; Dhyani, P.; Sharma, R.K. Spatial transcriptional dynamics of geographically separated genotypes revealed key regulators of podophyllotoxin biosynthesis in Podophyllum hexandrum. Ind. Crop. Prod. 2020, 147, 112247. [Google Scholar] [CrossRef]
- Singh, P.; Singh, G.; Bhandawat, A.; Singh, G.; Parmar, R.; Seth, R.; Sharma, R.K. Spatial transcriptome analysis provides insights of key gene (s) involved in steroidal saponin biosynthesis in medicinally important herb Trillium govanianum. Sci. Res. 2017, 7, 45295. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Bhandawat, A.; Parmar, R.; Singh, P.; Kumar, S.; Sharma, R.K. Global transcriptional insights of pollen-pistil interactions commencing self-incompatibility and fertilization in tea [Camellia sinensis (L.) O. Kuntze]. Int. J. Mol. Sci. 2019, 20, 539. [Google Scholar] [CrossRef] [PubMed]
- Maritim, T.K.; Seth, R.; Parmar, R.; Sharma, R.K. Multiple-genotypes transcriptional analysis revealed candidate genes and nucleotide variants for improvement of quality characteristics in tea (Camellia sinensis (L.) O. Kuntze). Genomics 2021, 113, 305–316. [Google Scholar] [CrossRef]
- Seth, R.; Maritim, T.K.; Parmar, R.; Sharma, R.K. Underpinning the molecular programming attributing heat stress associated thermotolerance in tea (Camellia sinensis (L.) O. Kuntze). Hortic. Res. 2021, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Smetanska, I. Production of secondary metabolites using plant cell cultures. Food Biotechnol. 2008, 111, 187–228. [Google Scholar] [CrossRef]
- Gani, U.; Vishwakarma, R.A.; Misra, P. Membrane transporters: The key drivers of transport of secondary metabolites in plants. Plant Cell Rep. 2020, 40, 1–18. [Google Scholar] [CrossRef]
- Singh, G.; Singh, G.; Singh, P.; Parmar, R.; Paul, N.; Vashist, R.; Swarnkar, M.K.; Kumar, A. Molecular dissection of transcriptional reprogramming of steviol glycosides synthesis in leaf tissue during developmental phase transitions in Stevia rebaudiana Bert. Sci. Rep. 2017, 7, 11835. [Google Scholar] [CrossRef]
- Shih, M.L.; Morgan, J.A. Metabolic flux analysis of secondary metabolism in plants. Metab. Eng. Commun. 2020, 10, 00123. [Google Scholar] [CrossRef]
- Patra, B.; Schluttenhofer, C.; Wu, Y.; Pattanaik, S.; Yuan, L. Transcriptional regulation of secondary metabolite biosynthesis in plants. Biochim. Biophys. Acta 2013, 1829, 12361247. [Google Scholar] [CrossRef]
- Van Moerkercke, A.; Steensma, P.; Schweizer, F.; Pollier, J.; Gariboldi, I.; Payne, R.; Bossche, R.V.; Miettinen, K.; Espoz, J.; Purnama, P.C.; et al. The bHLH transcription factor BIS1 controls the iridoid branch of the monoterpenoid indole alkaloid pathway in Catharanthus roseus. Proc. Natl. Acad. Sci. USA 2015, 112, 8130–8135. [Google Scholar] [CrossRef] [PubMed]
- Schluttenhofer, C.; Yuan, L. Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol. 2015, 167, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Chahel, A.A.; Zeng, S.; Yousaf, Z.; Liao, Y.; Yang, Z.; Wei, X.; Ying, W. Plant-specific transcription factor LrTCP4 enhances secondary metabolite biosynthesis in Lycium ruthenicum hairy roots. Plant Cell Tissue Org. Cult. 2019, 136, 323–337. [Google Scholar] [CrossRef]
- Kayani, S.I.; Shen, Q.; Rahman, S.U.; Fu, X.; Li, Y.; Wang, C.; Hassani, D.; Tang, K. Transcriptional regulation of flavonoid biosynthesis in Artemisia annua by AaYABBY5. Hortic. Res. 2021, 8, 257. [Google Scholar] [CrossRef]
- Lv, Z.; Wang, Y.; Liu, Y.; Peng, B.; Zhang, L.; Tang, K.; Chen, W. The SPB-box transcription factor AaSPL2 positively regulates artemisinin biosynthesis in Artemisia annua L. Front. Plant Sci. 2019, 10, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turgut-Kara, N.; Cakir, O. Comparative phylogenetic analysis of phenylpropanoid metabolism genes of legume plants. Plant Omics 2015, 8, 55. [Google Scholar] [CrossRef]
- Vranová, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef]
- Yang, L.; Ding, G.; Lin, H.; Cheng, H.; Kong, Y.; Wei, Y.; Fang, X.; Liu, R.; Wang, L.; Chen, X.; et al. Transcriptome analysis of medicinal plant Salvia miltiorrhiza and identification of genes related to tanshinone biosynthesis. PLoS ONE 2013, 8, e80464. [Google Scholar] [CrossRef]
- Devi, K.; Mishra, S.K.; Sahu, J.; Panda, D.; Modi, M.K.; Sen, P. Genome wide transcriptome profiling reveals differential gene expression in secondary metabolite pathway of Cymbopogon winterianus. Sci. Rep. 2016, 6, 21026. [Google Scholar] [CrossRef]
- Regueira, T.B.; Kildegaard, K.R.; Hansen, B.G.; Mortensen, U.H.; Hertweck, C.; Nielsen, J. Molecular basis for mycophenolic acid biosynthesis in Penicillium brevicompactum. Appl. Environ. Microbiol. 2011, 77, 3035–3043. [Google Scholar] [CrossRef]
- Sanchez, J.F.; Entwistle, R.; Corcoran, D.; Oakley, B.R.; Wang, C.C. Identification and molecular genetic analysis of the cichorine gene cluster in Aspergillus nidulans. MedChemComm 2012, 3, 997–1002. [Google Scholar] [CrossRef]
- Yaegashi, J.; Praseuth, M.B.; Tyan, S.W.; Sanchez, J.F.; Entwistle, R.; Chiang, Y.M.; Oakley, B.R.; Wang, C.C. Molecular genetic characterization of the biosynthesis cluster of a prenylated isoindolinone alkaloid aspernidine A in Aspergillus nidulans. Org. Lett. 2013, 15, 2862–2865. [Google Scholar] [CrossRef] [PubMed]
- Yonekura-Sakakibara, K.; Hanada, K. An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J. 2011, 66, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Seki, H.; Tamura, K.; Muranaka, T. P450s and UGTs: Key players in the structural diversity of triterpenoid saponins. Plant Cell Physiol. 2015, 56, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Yamada, Y.; Purwanto, R.; Minakuchi, Y.; Toyoda, A.; Hirakawa, H.; Sato, F. Mining of the uncharacterized cytochrome P450 genes involved in alkaloid biosynthesis in California poppy using a draft genome sequence. Plant Cell Physiol. 2018, 59, 222–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Zhang, J.; Liu, X.; Meng, M.; Wang, J.; Lin, J. Tissue-specific transcriptome for Dendrobium officinale reveals genes involved in flavonoid biosynthesis. Genomics 2020, 112, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Morant, M.; Schoch, G.A.; Ullmann, P.; Ertunç, T.; Little, D.; Olsen, C.E.; Petersen, M.; Negrel, J.; Werck-Reichhart, D. Catalytic activity, duplication and evolution of the CYP98 cytochrome P450 family in wheat. Plant Mol. Biol. 2007, 63, 1–19. [Google Scholar] [CrossRef]
- Cui, H.; Yu, X.; Wang, Y.; Cui, Y.; Li, X.; Liu, Z.; Qin, S. Evolutionary origins, molecular cloning and expression of carotenoid hydroxylases in eukaryotic photosynthetic algae. BMC Genom. 2013, 14, 457. [Google Scholar] [CrossRef]
- Magome, H.; Nomura, T.; Hanada, A.; Takeda-Kamiya, N.; Ohnishi, T.; Shinma, Y.; Katsumata, T.; Kawaide, H.; Kamiya, Y.; Yamaguchi, S. 2013. CYP714B1 and CYP714B2 encode gibberellin 13-oxidases that reduce gibberellin activity in rice. Proc. Natl. Acad. Sci. USA 2013, 110, 1947–1952. [Google Scholar] [CrossRef]
- Lin, J.S.; Huang, X.X.; Li, Q.; Cao, Y.; Bao, Y.; Meng, X.F.; Li, Y.J.; Fu, C.; Hou, B.K. UDP-glycosyltransferase 72B1 catalyzes the glucose conjugation of monolignols and is essential for the normal cell wall lignification in Arabidopsis thaliana. Plant J. 2016, 88, 26–42. [Google Scholar] [CrossRef]
- Irmler, S.; Schröder, G.; St-Pierre, B.; Crouch, N.P.; Hotze, M.; Schmidt, J.; Strack, D.; Matern, U.; Schröder, J. Indole alkaloid biosynthesis in Catharanthus roseus: New enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J. 2000, 24, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Jun, X.U.; Wang, X.Y.; Guo, W.Z. The cytochrome P450 superfamily: Key players in plant development and defense. J. Integr. Agric. 2015, 14, 1673–1686. [Google Scholar] [CrossRef]
- Fu, X.; Shi, P.; He, Q.; Shen, Q.; Tang, Y.; Pan, Q.; Ma, Y.; Yan, T.; Chen, M.; Hao, X.; et al. AaPDR3, a PDR transporter 3, is involved in sesquiterpene β-caryophyllene transport in Artemisia annua. Front. Plant Sci. 2017, 8, 723. [Google Scholar] [CrossRef]
- Ghawana, S.; Paul, A.; Kumar, H.; Kumar, A.; Singh, H.; Bhardwaj, P.K.; Rani, A.; Singh, R.S.; Raizada, J.; Singh, K.; et al. An RNA isolation system for plant tissues rich in secondary metabolites. BMC Res. Notes 2011, 4, 85. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, 64–70. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, 447–452. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.Y. ReactomePA:an R/Bioconductor package for reactome pathway analysis and visualization. Mol. BioSyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Walia, S.; Kumar, R. Elucidating the yield and quality response of Tagetes minuta L. intercropped with Zea mays L. under different spacing in the western Himalayas. Ind. Crop. Prod. 2021, 171, 113850. [Google Scholar] [CrossRef]
- Dadwal, V.; Agrawal, H.; Sonkhla, K.; Joshi, R.; Gupta, M. Characterization of phenolics, amino acids, fatty acids and antioxidant activity in pulp and seeds of high altitude Himalayan crab apple fruits (Malus baccata). Food Sci. Technol. 2018, 55, 2160–2169. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Sample Name | Raw Reads | Filtered Reads | Retained High-Quality Reads (%) |
|---|---|---|---|---|
| 1 | Leaf | 43,893,016 | 40,144,376 | 91.4 |
| 2 | Stem | 39,814,592 | 36,737,576 | 92.2 |
| 3 | Root | 35,495,806 | 32,757,628 | 92.2 |
| Total reads | 119,203,414 | 109,639,580 | 91.9 |
| De Novo-Assembly Statistics | ||
|---|---|---|
| Before Clustering | After Clustering | |
| Total transcripts | 1,61,836 | 1,17,214 |
| Total genes | 81,570 | 81,162 |
| Percent GC | 39.69 | 39.62 |
| Contig N50 | 1751 | 1607 bp |
| Median contig length | 715 | 526 bp |
| Average contig length | 1071.45 | 923.32 bp |
| Database | No. of Transcripts | Percent (%) |
|---|---|---|
| NCBI nr | 69,731 | 59.49 |
| TAIR | 58,982 | 50.31 |
| KOG | 55,513 | 47.36 |
| Swiss-Prot | 53,957 | 46.03 |
| Plant TFDB | 33,040 | 28.18 |
| KEGG | 8945 | 7.63 |
| Total | 72,120 | 61.53 |
| S. No. | TF Family | Leaf (L) | Stem (S) | Root (R) |
|---|---|---|---|---|
| 1 | bHLH | 65 | 47 | 19 |
| 2 | NAC | 33 | 54 | 16 |
| 3 | MYB related | 42 | 46 | 24 |
| 4 | ERF | 77 | 71 | 22 |
| 5 | WRKY | 18 | 20 | 4 |
| 6 | C2H2 | 20 | 21 | 9 |
| 7 | B3 | 33 | 14 | 9 |
| 8 | C3H | 16 | 23 | 2 |
| 9 | FAR1 | 21 | 16 | 14 |
| 10 | MYB | 17 | 21 | 5 |
| S. No. | District | Population | Latitude (°N) | Longitude (°E) | Altitude (m) |
|---|---|---|---|---|---|
| 1 | Kinnaur | Asrang (G1) | 31.667 | 78.313 | 3376 |
| 2 | Kullu | Kandhaghai (G2) | 31.408 | 77.444 | 2250 |
| 3 | Mandi | Thunag (G3) | 31.577 | 77.152 | 2177 |
| 4 | Kullu | Gulaba (G4) | 32.317 | 77.189 | 2521 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devi, A.; Seth, R.; Masand, M.; Singh, G.; Holkar, A.; Sharma, S.; Singh, A.; Sharma, R.K. Spatial Genomic Resource Reveals Molecular Insights into Key Bioactive-Metabolite Biosynthesis in Endangered Angelica glauca Edgew. Int. J. Mol. Sci. 2022, 23, 11064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911064
Devi A, Seth R, Masand M, Singh G, Holkar A, Sharma S, Singh A, Sharma RK. Spatial Genomic Resource Reveals Molecular Insights into Key Bioactive-Metabolite Biosynthesis in Endangered Angelica glauca Edgew. International Journal of Molecular Sciences. 2022; 23(19):11064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911064
Chicago/Turabian StyleDevi, Amna, Romit Seth, Mamta Masand, Gopal Singh, Ashlesha Holkar, Shikha Sharma, Ashok Singh, and Ram Kumar Sharma. 2022. "Spatial Genomic Resource Reveals Molecular Insights into Key Bioactive-Metabolite Biosynthesis in Endangered Angelica glauca Edgew" International Journal of Molecular Sciences 23, no. 19: 11064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911064