

Characterizing Antimicrobial Resistance in Clinically Relevant Bacteria Isolated at the Human/Animal/Environment Interface Using Whole-Genome Sequencing in Austria
 , , , , , ,
, , , , , ,
Abstract
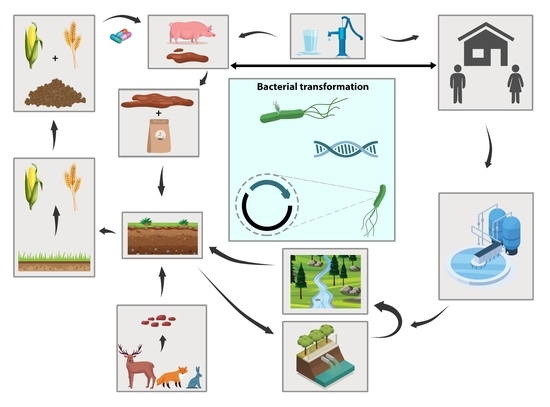
:
1. Introduction
2. Results
2.1. Isolates by Species and Compartment
2.2. Phylogenetic Relationships
2.3. Phenotypic AMR
2.4. ARGs, VGs and Plasmids
3. Discussion
4. Materials and Methods
4.1. Sampling
4.2. Isolate Selection and Sequencing
4.3. Whole Genome Sequencing and Typing
4.4. Phenotypic Resistance
4.5. Sequence Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Robinson, T.P.; Bu, D.P.; Carrique-Mas, J.; Fèvre, E.M.; Gilbert, M.; Grace, D.; Hay, S.I.; Jiwakanon, J.; Kakkar, M.; Kariuki, S.; et al. Antibiotic resistance is the quintessential One Health issue. Trans. R. Trop. Med. Hyg. 2016, 110, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis 2019, 19, 56–66. [Google Scholar] [CrossRef]
- Apfalter, P.; Berning, L.; Eigentler, A.; El-Belazi, G.; Fellinger, F.; Klemens, F.; Fuchs, R.; Hain, C.; Hartl, R.; Igler, G.; et al. Resistenzbericht Österreich AURES; Bundesministerium für Soziales, Gesundheit, Pflege und Konsumentenschutz (BMSGPK): Wien, Austria, 2020. [Google Scholar]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Khurshid, M.; Arshad, M.I.; Muzammil, S.; Rasool, M.; Yasmeen, N.; Shah, T.; Chaudhry, T.H.; Rasool, M.H.; Shahid, A.; et al. Antibiotic Resistance: One Health One World Outlook. Front. Cell. Infect. Microbiol. 2021, 11, 1153. [Google Scholar] [CrossRef]
- Destoumieux-Garzón, D.; Mavingui, P.; Boetsch, G.; Boissier, J.; Darriet, F.; Duboz, P.; Fritsch, C.; Giraudoux, P.; Le Roux, F.; Morand, S.; et al. The One Health Concept: 10 Years Old and a Long Road Ahead. Front. Vet. Sci. 2018, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Calistri, P.; Iannetti, S.; Danzetta, M.L.; Narcisi, V.; Cito, F.; Di Sabatino, D.; Bruno, R.; Sauro, F.; Atzeni, M.; Carvelli, A.; et al. The Components of ‘One World—One Health’ Approach. Transbound. Emerg. Dis. 2013, 60, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, P.M.C.; Flach, C.-F.; Larsson, D.G.J. A conceptual framework for the environmental surveillance of antibiotics and antibiotic resistance. Environ. Int. 2019, 130, 104880. [Google Scholar] [CrossRef]
- Dolejska, M.; Literak, I. Wildlife Is Overlooked in the Epidemiology of Medically Important Antibiotic-Resistant Bacteria. Antimicrob. Agents Chemother. 2019, 63, e01167-19. [Google Scholar] [CrossRef] [Green Version]
- Jechalke, S.; Schierstaedt, J.; Becker, M.; Flemer, B.; Grosch, R.; Smalla, K.; Schikora, A. Salmonella Establishment in Agricultural Soil and Colonization of Crop Plants Depend on Soil Type and Plant Species. Front. Microbiol. 2019, 10, 967. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.A.; Curriero, F.C.; Cosgrove, S.E.; Nachman, K.E.; Schwartz, B.S. High-density livestock operations, crop field application of manure, and risk of community-associated methicillin-resistant Staphylococcus aureus infection in Pennsylvania. JAMA Intern. Med. 2013, 173, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Kalupahana, R.S.; Duim, B.; Verstappen, K.M.; Gamage, C.D.; Dissanayake, N.; Ranatunga, L.; Graveland, H.; Wagenaar, J.A. MRSA in Pigs and the Environment as a Risk for Employees in Pig-Dense Areas of Sri Lanka. Front. Sustain. Food Syst. 2019, 3, 967. [Google Scholar] [CrossRef]
- Radu, E.; Woegerbauer, M.; Rab, G.; Oismüller, M.; Strauss, P.; Hufnagl, P.; Gottsberger, R.A.; Krampe, J.; Weyermair, K.; Kreuzinger, N. Resilience of agricultural soils to antibiotic resistance genes introduced by agricultural management practices. Sci. Total Environ. 2021, 756, 143699. [Google Scholar] [CrossRef] [PubMed]
- Hölzel, C.S.; Tetens, J.L.; Schwaiger, K. Unraveling the Role of Vegetables in Spreading Antimicrobial-Resistant Bacteria: A Need for Quantitative Risk Assessment. Foodborne Pathog. Dis. 2018, 15, 671–688. [Google Scholar] [CrossRef]
- Matsukawa, M.; Igarashi, M.; Watanabe, H.; Qin, L.; Ohnishi, M.; Terajima, J.; Iyoda, S.; Morita-Ishihara, T.; Tateda, K.; Ishii, Y.; et al. Epidemiology and genotypic characterisation of dissemination patterns of uropathogenic Escherichia coli in a community. Epidemiol. Infect. 2019, 147, e148. [Google Scholar] [CrossRef] [PubMed]
- Manges, A.R.; Geum, H.M.; Guo, A.; Edens, T.J.; Fibke, C.D.; Pitout, J.D.D. Global Extraintestinal Pathogenic Escherichia coli (ExPEC) Lineages. Clin. Microbiol. Rev. 2019, 32, e00135-18. [Google Scholar] [CrossRef] [PubMed]
- Royer, G.; Darty, M.M.; Clermont, O.; Condamine, B.; Laouenan, C.; Decousser, J.-W.; Vallenet, D.; Lefort, A.; de Lastours, V.; Denamur, E.; et al. Phylogroup stability contrasts with high within sequence type complex dynamics of Escherichia coli bloodstream infection isolates over a 12-year period. Genome Med. 2021, 13, 77. [Google Scholar] [CrossRef]
- Tetzschner, A.M.M.; Johnson, J.R.; Johnston, B.D.; Lund, O.; Scheutz, F.; Dekker, J.P. In Silico Genotyping of Escherichia coli Isolates for Extraintestinal Virulence Genes by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol 2020, 58, e01269-20. [Google Scholar] [CrossRef]
- De Carvalho, M.P.N.; Fernandes, M.R.; Sellera, F.P.; Lopes, R.; Monte, D.F.; Hippólito, A.G.; Milanelo, L.; Raso, T.F.; Lincopan, N. International clones of extended-spectrum β-lactamase (CTX-M)-producing Escherichia coli in peri-urban wild animals, Brazil. Transbound. Emerg. Dis. 2020, 67, 1804–1815. [Google Scholar] [CrossRef] [Green Version]
- Decano, A.G.; Pettigrew, K.; Sabiiti, W.; Sloan, D.J.; Neema, S.; Bazira, J.; Kiiru, J.; Onyango, H.; Asiimwe, B.; Holden, M.T.G. Pan-Resistome Characterization of Uropathogenic Escherichia coli and Klebsiella pneumoniae Strains Circulating in Uganda and Kenya, Isolated from 2017–2018. Antibiotics 2021, 10, 1547. [Google Scholar] [CrossRef] [PubMed]
- Tarabai, H.; Wyrsch, E.R.; Bitar, I.; Dolejska, M.; Djordjevic, S.P. Epidemic HI2 Plasmids Mobilising the Carbapenemase Gene blaIMP-4 in Australian Clinical Samples Identified in Multiple Sublineages of Escherichia coli ST216 Colonising Silver Gulls. Microorganisms 2021, 9, 567. [Google Scholar] [CrossRef]
- Loong, S.K.; Mahfodz, N.H.; Che Mat Seri, N.A.; Mohamad Wali, H.A.; Abd Gani, S.A.; Wong, P.F.; AbuBakar, S. Genetic characterization of commensal Escherichia coli isolated from laboratory rodents. Springerplus 2016, 5, 1035. [Google Scholar] [CrossRef] [PubMed]
- Valenza, G.; Werner, M.; Eisenberger, D.; Nickel, S.; Lehner-Reindl, V.; Höller, C.; Bogdan, C. First report of the new emerging global clone ST1193 among clinical isolates of extended-spectrum β-lactamase (ESBL)-producing Escherichia coli from Germany. J. Glob. Antimicrob. Resist. 2019, 17, 305–308. [Google Scholar] [CrossRef]
- Cabal, A.; Peischl, N.; Rab, G.; Stöger, A.; Springer, B.; Sucher, J.; Allerberger, F.; Ruppitsch, W. Draft Genome Sequence of a Multidrug-Resistant Escherichia coli Sequence Type 1193 Pandemic Clone Isolated from Wastewater in Austria. Microbiol. Resour. Announc. 2021, 10, e0076221. [Google Scholar] [CrossRef] [PubMed]
- Doumith, M.; Day, M.; Ciesielczuk, H.; Hope, R.; Underwood, A.; Reynolds, R.; Wain, J.; Livermore, D.M.; Woodford, N. Rapid identification of major Escherichia coli sequence types causing urinary tract and bloodstream infections. J. Clin. Microbiol. 2015, 53, 160–166. [Google Scholar] [CrossRef]
- Kieffer, N.; Royer, G.; Decousser, J.-W.; Bourrel, A.-S.; Palmieri, M.; Rosa, J.-M.O.D.L.; Jacquier, H.; Denamur, E.; Nordmann, P.; Poirel, L. mcr-9, an Inducible Gene Encoding an Acquired Phosphoethanolamine Transferase in Escherichia coli, and Its Origin. Antimicrob. Agents Chemother. 2019, 63, e00965-19. [Google Scholar] [CrossRef]
- Nobili, G.; Franconieri, I.; Basanisi, M.G.; La Bella, G.; Tozzoli, R.; Caprioli, A.; La Salandra, G. Short communication: Isolation of Shiga toxin-producing Escherichia coli in raw milk and mozzarella cheese in southern Italy. J. Dairy Sci. 2016, 99, 7877–7880. [Google Scholar] [CrossRef]
- Eggert, M.; StüBer, E.; Heurich, M.; Fredriksson-Ahomaa, M.; Burgos, Y.; Beutin, L.; Märtlbauer, E. Detection and characterization of Shiga toxin-producing Escherichia coli in faeces and lymphatic tissue of free-ranging deer. Epidemiol. Infect. 2013, 141, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Newton, H.J.; Sloan, J.; Bulach, D.M.; Seemann, T.; Allison, C.C.; Tauschek, M.; Robins-Browne, R.M.; Paton, J.C.; Whittam, T.S.; Paton, A.W.; et al. Shiga toxin-producing Escherichia coli strains negative for locus of enterocyte effacement. Emerg. Infect. Dis. 2009, 15, 372–380. [Google Scholar] [CrossRef]
- Söderlund, R.; Hurel, J.; Jinnerot, T.; Sekse, C.; Aspán, A.; Eriksson, E.; Bongcam-Rudloff, E. Genomic comparison of Escherichia coli serotype O103:H2 isolates with and without verotoxin genes: Implications for risk assessment of strains commonly found in ruminant reservoirs. Infect. Ecol. Epidemiol. 2016, 6, 30246. [Google Scholar] [CrossRef]
- Hernandes, R.T.; Elias, W.P.; Vieira, M.A.M.; Gomes, T.A.T. An overview of atypical enteropathogenic Escherichia coli. FEMS Microbiol. Lett. 2009, 297, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Paterson, D.L. Detection of plasmid-mediated class C β-lactamases. Int. J. Infect. Dis. 2007, 11, 191–197. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Use of Aminoglycosides in Animals in the European Union: Development of Resistance and Impact on Human and Animal Health; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2018.
- Awad, A.; Arafat, N.; Elhadidy, M. Genetic elements associated with antimicrobial resistance among avian pathogenic Escherichia coli. Ann. Clin. Microbiol. Antimicrob. 2016, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Norström, M. The genetic background for streptomycin resistance in Escherichia coli influences the distribution of MICs. J. Antimicrob. Chemother. 2005, 56, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, N.; Usui, M.; Fukuda, A.; Asai, T.; Higuchi, H.; Okamoto, E.; Seki, K.; Takada, H.; Tamura, Y. Manure Compost Is a Potential Source of Tetracycline-Resistant Escherichia coli and Tetracycline Resistance Genes in Japanese Farms. Antibiotics 2020, 9, 76. [Google Scholar] [CrossRef]
- Plaza-Rodríguez, C.; Alt, K.; Grobbel, M.; Hammerl, J.A.; Irrgang, A.; Szabo, I.; Stingl, K.; Schuh, E.; Wiehle, L.; Pfefferkorn, B.; et al. Wildlife as Sentinels of Antimicrobial Resistance in Germany? Front. Vet. Sci. 2020, 7, 627821. [Google Scholar] [CrossRef]
- Wareth, G.; Neubauer, H. The Animal-foods-environment interface of Klebsiella pneumoniae in Germany: An observational study on pathogenicity, resistance development and the current situation. Vet. Res. 2021, 52, 16. [Google Scholar] [CrossRef]
- Liu, L.; Feng, Y.; Wei, L.; Xiao, Y.; Zong, Z. KPC-2-Producing Carbapenem-Resistant Klebsiella pneumoniae of the Uncommon ST29 Type Carrying OXA-926, a Novel Narrow-Spectrum OXA β-Lactamase. Front. Microbiol. 2021, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Wyres, K.L.; Holt, K.E. Klebsiella pneumoniae as a key trafficker of drug resistance genes from environmental to clinically important bacteria. Curr. Opin. Microbiol. 2018, 45, 131–139. [Google Scholar] [CrossRef]
- Heinz, E.; Ejaz, H.; Bartholdson Scott, J.; Wang, N.; Gujaran, S.; Pickard, D.; Wilksch, J.; Cao, H.; Haq, I.-U.; Dougan, G.; et al. Resistance mechanisms and population structure of highly drug resistant Klebsiella in Pakistan during the introduction of the carbapenemase NDM-1. Sci. Rep. 2019, 9, 2392. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-Y.; Lu, P.-L.; Tseng, S.-P. Update on fosfomycin-modified genes in Enterobacteriaceae. J. Microbiol. Immunol. Infect. 2019, 52, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xu, X.; Guo, Q.; Wang, P.; Wang, W.; Wang, M. Characterization of fosA5, a new plasmid-mediated fosfomycin resistance gene in Escherichia coli. Lett. Appl. Microbiol. 2015, 60, 259–264. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Chen, Y.-H.; Hung, I.-C.; Chu, P.-H.; Chang, Y.-H.; Lin, Y.-T.; Yang, H.-C.; Wang, J.-T. Transporter Genes and fosA Associated With Fosfomycin Resistance in Carbapenem-Resistant Klebsiella pneumoniae. Front. Microbiol. 2022, 13. [Google Scholar] [CrossRef]
- Cubero, M.; Grau, I.; Tubau, F.; Pallarés, R.; Dominguez, M.A.; Liñares, J.; Ardanuy, C. Hypervirulent Klebsiella pneumoniae clones causing bacteraemia in adults in a teaching hospital in Barcelona, Spain (2007–2013). Clin. Microbiol. Infect. 2016, 22, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Patro, L.P.P.; Sudhakar, K.U.; Rathinavelan, T. K-PAM: A unified platform to distinguish Klebsiella species K- and O-antigen types, model antigen structures and identify hypervirulent strains. Sci. Rep. 2020, 10, 16732. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, T.; Chen, L.; Du, H. Virulence Factors in Hypervirulent Klebsiella pneumoniae. Front. Microbiol. 2021, 12, 642484. [Google Scholar] [CrossRef]
- Lam, M.M.C.; Wick, R.R.; Watts, S.C.; Cerdeira, L.T.; Wyres, K.L.; Holt, K.E. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 2021, 12, 4188. [Google Scholar] [CrossRef]
- Aun, E.; Kisand, V.; Laht, M.; Telling, K.; Kalmus, P.; Väli, Ü.; Brauer, A.; Remm, M.; Tenson, T. Molecular Characterization of Enterococcus Isolates From Different Sources in Estonia Reveals Potential Transmission of Resistance Genes Among Different Reservoirs. Front. Microbiol. 2021, 12, 601490. [Google Scholar] [CrossRef]
- Hollenbeck, B.L.; Rice, L.B. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 2012, 3, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Rathnayake, I.; Hargreaves, M.; Huygens, F. SNP diversity of Enterococcus faecalis and Enterococcus faecium in a South East Queensland waterway, Australia, and associated antibiotic resistance gene profiles. BMC Microbiol. 2011, 11, 201. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, M.; Bogaerts, B.; Vanneste, K.; De Keersmaecker, S.C.J.; Roosens, N.H.C.; Kowalewicz, C.; Simon, G.; Argudín, M.A.; Deplano, A.; Hallin, M.; et al. Large diversity of linezolid-resistant isolates discovered in food-producing animals through linezolid selective monitoring in Belgium in 2019. J. Antimicrob. Chemother. 2021, 77, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Jordan, D.; Sahibzada, S.; Abraham, R.; Pang, S.; Coombs, G.W.; O’Dea, M.; Abraham, S. Antimicrobial Resistance in Porcine Enterococci in Australia and the Ramifications for Human Health. Appl. Environ. Microbiol 2021, 87, e03037-20. [Google Scholar] [CrossRef]
- Pöntinen, A.K.; Top, J.; Arredondo-Alonso, S.; Tonkin-Hill, G.; Freitas, A.R.; Novais, C.; Gladstone, R.A.; Pesonen, M.; Meneses, R.; Pesonen, H.; et al. Apparent nosocomial adaptation of Enterococcus faecalis predates the modern hospital era. Nat. Commun. 2021, 12, 1523. [Google Scholar] [CrossRef]
- León-Sampedro, R.; Del Campo, R.; Rodriguez-Baños, M.; Lanza, V.F.; Pozuelo, M.J.; Francés-Cuesta, C.; Tedim, A.P.; Freitas, A.R.; Novais, C.; Peixe, L.; et al. Phylogenomics of Enterococcus faecalis from wild birds: New insights into host-associated differences in core and accessory genomes of the species. Environ. Microbiol. 2019, 21, 3046–3062. [Google Scholar] [CrossRef] [PubMed]
- Chilambi, G.S.; Nordstrom, H.R.; Evans, D.R.; Kowalski, R.P.; Dhaliwal, D.K.; Jhanji, V.; Shanks, R.M.Q.; Van Tyne, D. Genomic and phenotypic diversity of Enterococcus faecalis isolated from endophthalmitis. PLoS ONE 2021, 16, e0250084. [Google Scholar] [CrossRef]
- Holman, D.B.; Klima, C.L.; Gzyl, K.E.; Zaheer, R.; Service, C.; Jones, T.H.; McAllister, T.A. Antimicrobial Resistance in Enterococcus Spp. Isolated from a Beef Processing Plant and Retail Ground Beef. Microbiol. Spectr. 2021, 9, e0198021. [Google Scholar] [CrossRef]
- Zhu, X.Q.; Wang, X.M.; Li, H.; Shang, Y.H.; Pan, Y.S.; Wu, C.M.; Wang, Y.; Du, X.D.; Shen, J.Z. Novel lnu(G) gene conferring resistance to lincomycin by nucleotidylation, located on Tn6260 from Enterococcus faecalis E531. J. Antimicrob. Chemother. 2017, 72, 993–997. [Google Scholar] [CrossRef]
- Li, L.; Sun, J.; Liu, B.; Zhao, D.; Ma, J.; Deng, H.; Li, X.; Hu, F.; Liao, X.; Liu, Y. Quantification of lincomycin resistance genes associated with lincomycin residues in waters and soils adjacent to representative swine farms in China. Front. Microbiol. 2013, 4, 364. [Google Scholar] [CrossRef]
- Coque, T.M.; Singh, K.V.; Weinstock, G.M.; Murray, B.E. Characterization of dihydrofolate reductase genes from trimethoprim-susceptible and trimethoprim-resistant strains of Enterococcus faecalis. Antimicrob. Agents Chemother. 1999, 43, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Zaheer, R.; Cook, S.R.; Barbieri, R.; Goji, N.; Cameron, A.; Petkau, A.; Polo, R.O.; Tymensen, L.; Stamm, C.; Song, J.; et al. Surveillance of Enterococcus spp. reveals distinct species and antimicrobial resistance diversity across a One-Health continuum. Sci. Rep. 2020, 10, 3937. [Google Scholar] [CrossRef] [PubMed]
- Diab, M.; Salem, D.; El-Shenawy, A.; El-Far, A.; Abdelghany, A.; Awad, A.R.; El Defrawy, I.; Shemis, M. Detection of high level aminoglycoside resistance genes among clinical isolates of Enterococcus species. Egypt. J. Med. Hum. Genet. 2019, 20, 28. [Google Scholar] [CrossRef]
- Akhtar, M.; Hirt, H.; Zurek, L. Horizontal transfer of the tetracycline resistance gene tetM mediated by pCF10 among Enterococcus faecalis in the house fly (Musca domestica L.) alimentary canal. Microb. Ecol. 2009, 58, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.M.; Talavera, G.N.; Hernández, L.A.R.; Weisberg, S.B.; Ambrose, R.F.; Jay, J.A. Virulence Genes among Enterococcus faecalis and Enterococcus faecium Isolated from Coastal Beaches and Human and Nonhuman Sources in Southern California and Puerto Rico. J. Pathog. 2016, 2016, 3437214. [Google Scholar] [CrossRef]
- Bakshi, U.; Sarkar, M.; Paul, S.; Dutta, C. Assessment of virulence potential of uncharacterized Enterococcus faecalis strains using pan genomic approach—Identification of pathogen–specific and habitat-specific genes. Sci. Rep. 2016, 6, 38648. [Google Scholar] [CrossRef]
- Blöschl, G.; Blaschke, A.P.; Broer, M.; Bucher, C.; Carr, G.; Chen, X.; Eder, A.; Exner-Kittridge, M.; Farnleitner, A.; Flores-Orozco, A.; et al. The Hydrological Open Air Laboratory (HOAL) in Petzenkirchen: A hypothesis-driven observatory. Hydrol. Earth Syst. Sci. 2016, 20, 227–255. [Google Scholar] [CrossRef]
- Pietzka, A.; Murer, A.; Lennkh, A.; Hauser, K.; Vötsch, K.; Springer, B.; Allerberger, F.; Ruppitsch, W. Draft Genome Sequences of Two Listeria monocytogenes Strains Isolated from Invasive Snails (Arion vulgaris) in Austria in 2019. Microbiol. Resour. Announc. 2021, 10, e0037521. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 September 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Jünemann, S.; Sedlazeck, F.J.; Prior, K.; Albersmeier, A.; John, U.; Kalinowski, J.; Mellmann, A.; Goesmann, A.; Von Haeseler, A.; Stoye, J.; et al. Updating benchtop sequencing performance comparison. Nat. Biotechnol. 2013, 31, 294–296. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Prior, K.; Bender, J.K.; Harmsen, D.; Klare, I.; Fuchs, S.; Bethe, A.; Zühlke, D.; Göhler, A.; Schwarz, S.; et al. A Core Genome Multilocus Sequence Typing Scheme for Enterococcus faecalis. J. Clin. Microbiol. 2019, 57, e01686-18. [Google Scholar] [CrossRef] [PubMed]
- de Been, M.; Pinholt, M.; Top, J.; Bletz, S.; Mellmann, A.; van Schaik, W.; Brouwer, E.; Rogers, M.; Kraat, Y.; Bonten, M.; et al. Core Genome Multilocus Sequence Typing Scheme for High- Resolution Typing of Enterococcus faecium. J. Clin. Microbiol. 2015, 53, 3788–3797. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019, 48, D517–D525. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2018, 47, D687–D692. [Google Scholar] [CrossRef]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods Mol. Biol. 2020, 2075, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Heinz, E.; Holt, K.E.; Wyres, K.L. Kaptive Web: User-Friendly Capsule and Lipopolysaccharide Serotype Prediction for Klebsiella Genomes. J. Clin. Microbiol. 2018, 56, e00197-18. [Google Scholar] [CrossRef]
- Waters, N.R.; Abram, F.; Brennan, F.; Holmes, A.; Pritchard, L. Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. 2020, 2, acmi000143. [Google Scholar] [CrossRef]
- Joensen, K.G.; Tetzschner, A.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and Easy In Silico Serotyping of Escherichia coli Isolates by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef] [Green Version]
- EFSA (European Food Safety Authority); Aerts, M.; Battisti, A.; Hendriksen, R.; Kempf, I.; Teale, C.; Tenhagen, B.-A.; Veldman, K.; Wasyl, D.; Guerra, B.; et al. Technical specifications on harmonised monitoring of antimicrobial resistance in zoonotic and indicator bacteria from food-producing animals and food. EFSA J. 2019, 17, e05709. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Feed | Field Drainage | Groundwater | Pig Manure | River | Soil | Wastewater | Wildlife | Crops | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| E. coli | 1 | 5 | 2 | 1 | 5 | 4 | 12 | 19 | 0 | 49 |
| E. faecalis | 0 | 0 | 1 | 5 | 2 | 3 | 6 | 10 | 0 | 27 |
| K. pneumoniae | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 7 |
| E. faecium | 0 | 0 | 3 | 0 | 0 | 0 | 3 | 0 | 0 | 6 |
| Total | 1 | 5 | 7 | 5 | 7 | 7 | 28 | 29 | 0 | 89 |
| Species | AMR Pattern | Isolates, n (%) | |
|---|---|---|---|
| E. coli | AMP/AZT*/CEP/CIP/CTX/ERY*/GEN/MOX*/ STR*/TS* | 1 | 9/49 (18.3%) |
| AMP/CIP/MOX*/STR*/TET*/TS* | 2 | ||
| AMC/AMP | 1 | ||
| STR*/TET* | 2 | ||
| AMP | 1 | ||
| TET* | 2 | ||
| K. pneumoniae | AMP/FOS* | 5 | 7/7 (100%) |
| AMP | 2 | ||
| E. faecium | AMP | 1 | 1/6 (16.7%) |
| E. faecalis | KAN*/STR*/TET* | 2 | 9/24 (33.3%) |
| ERY*/TET* | 1 | ||
| CLY*/TET* | 1 | ||
| TET* | 5 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabal, A.; Rab, G.; Daza-Prieto, B.; Stöger, A.; Peischl, N.; Chakeri, A.; Mo, S.S.; Bock, H.; Fuchs, K.; Sucher, J.; et al. Characterizing Antimicrobial Resistance in Clinically Relevant Bacteria Isolated at the Human/Animal/Environment Interface Using Whole-Genome Sequencing in Austria. Int. J. Mol. Sci. 2022, 23, 11276. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911276
Cabal A, Rab G, Daza-Prieto B, Stöger A, Peischl N, Chakeri A, Mo SS, Bock H, Fuchs K, Sucher J, et al. Characterizing Antimicrobial Resistance in Clinically Relevant Bacteria Isolated at the Human/Animal/Environment Interface Using Whole-Genome Sequencing in Austria. International Journal of Molecular Sciences. 2022; 23(19):11276. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911276
Chicago/Turabian StyleCabal, Adriana, Gerhard Rab, Beatriz Daza-Prieto, Anna Stöger, Nadine Peischl, Ali Chakeri, Solveig Sølverød Mo, Harald Bock, Klemens Fuchs, Jasmin Sucher, and et al. 2022. "Characterizing Antimicrobial Resistance in Clinically Relevant Bacteria Isolated at the Human/Animal/Environment Interface Using Whole-Genome Sequencing in Austria" International Journal of Molecular Sciences 23, no. 19: 11276. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911276