
Pro-Apoptotic and Anti-Invasive Properties Underscore the Tumor-Suppressing Impact of Myoglobin on a Subset of Human Breast Cancer Cells

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Myoglobin Impacts Breast Cancer Cell Survival
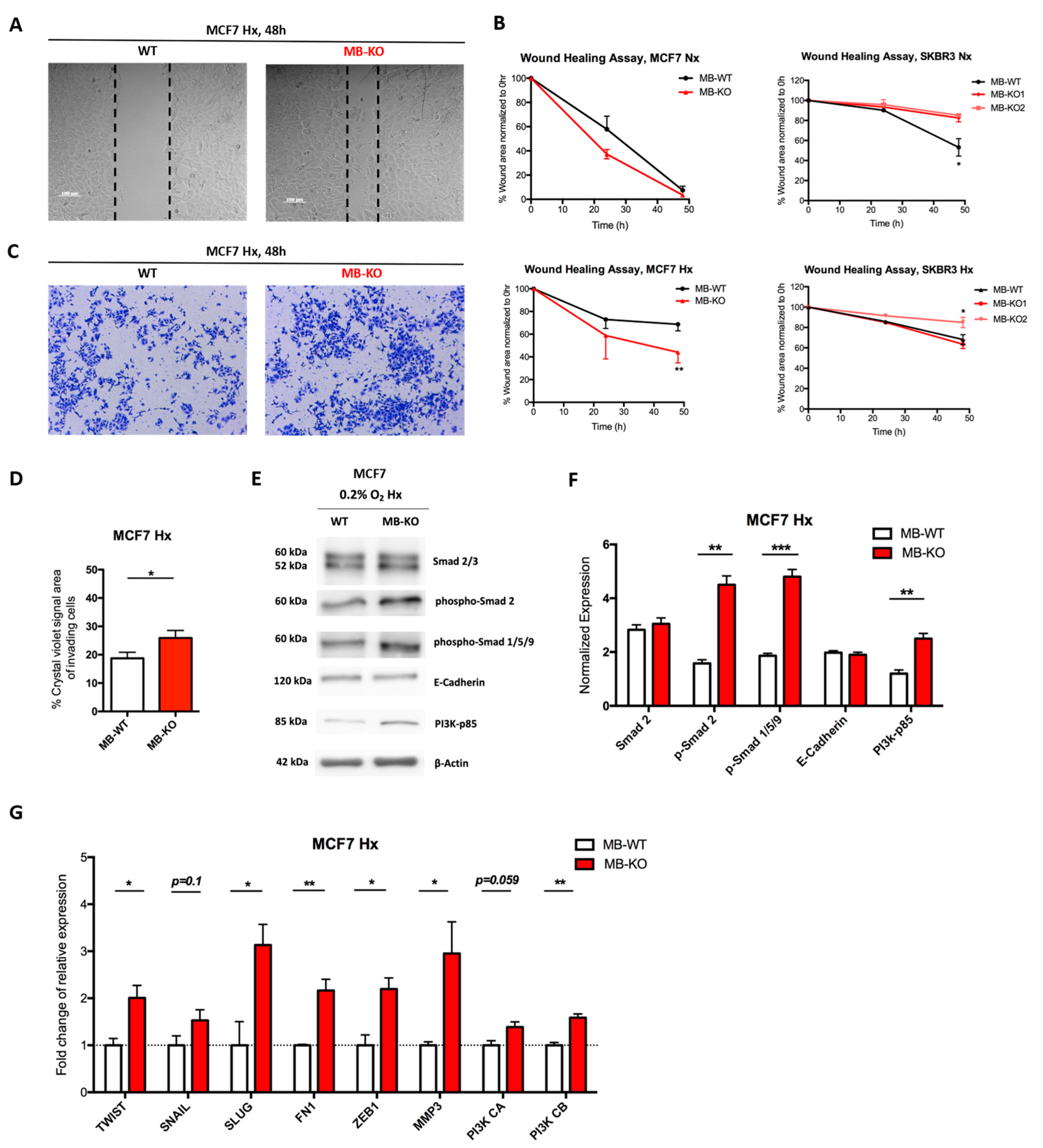
2.2. Loss of Myoglobin Increases the Migratory Capacity of Hypoxic MCF7 Cancer Cells
2.3. Endogenously Expressed Myoglobin in BrCa Cells Has No Impact on Cellular Response to Hypoxia
2.4. Myoglobin Interferes with Estrogen Receptor Expression and ROS Generation
2.5. Myoglobin Interferes with Cancer Cells’ Response to Chemotherapeutic but Not Ionizing Irradiation Treatment
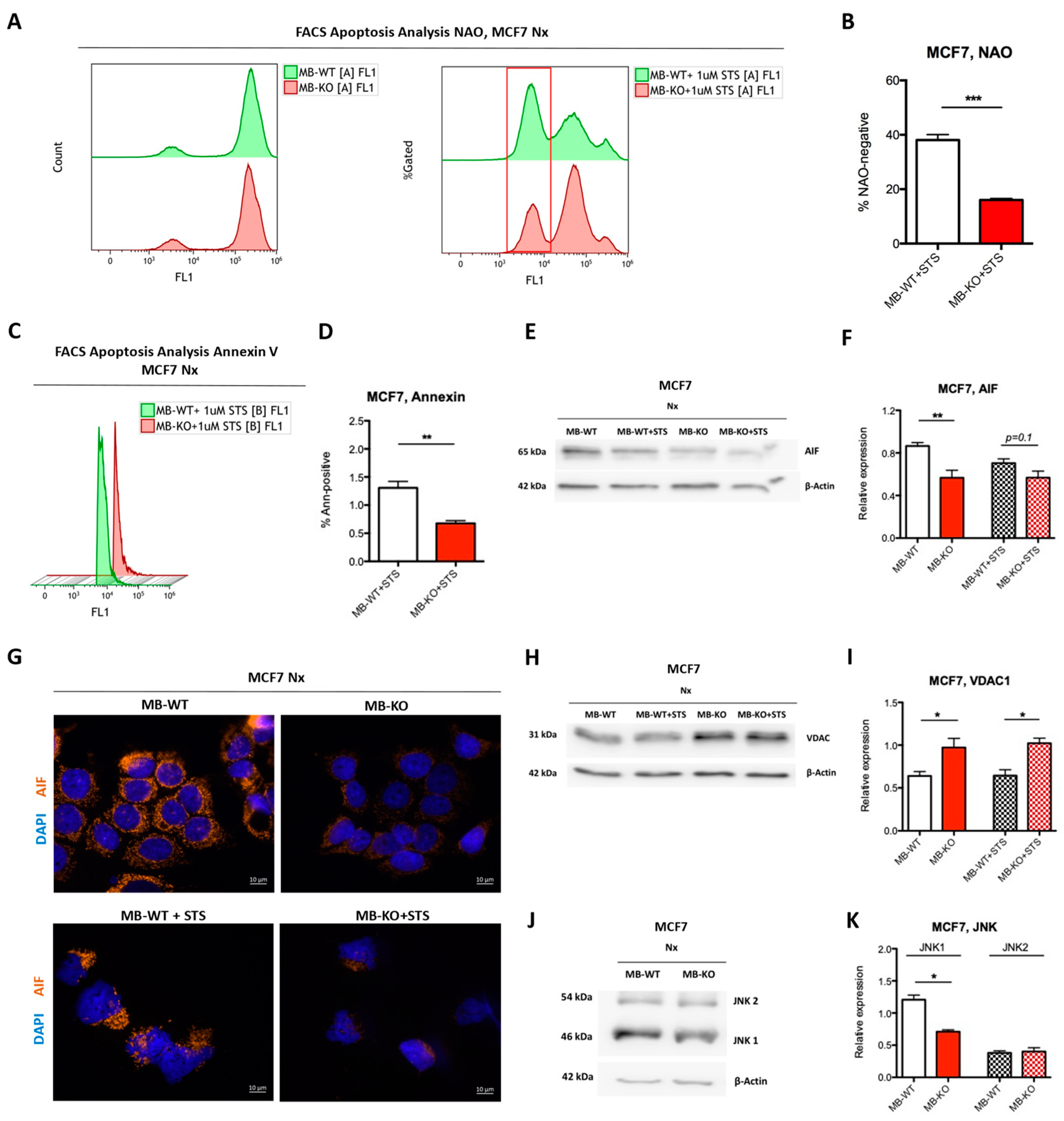
2.6. Loss of Myoglobin Inhibits Apoptosis in Normoxic BrCa Cells
2.7. Differential Gene Expression Analysis by RNA-Seq
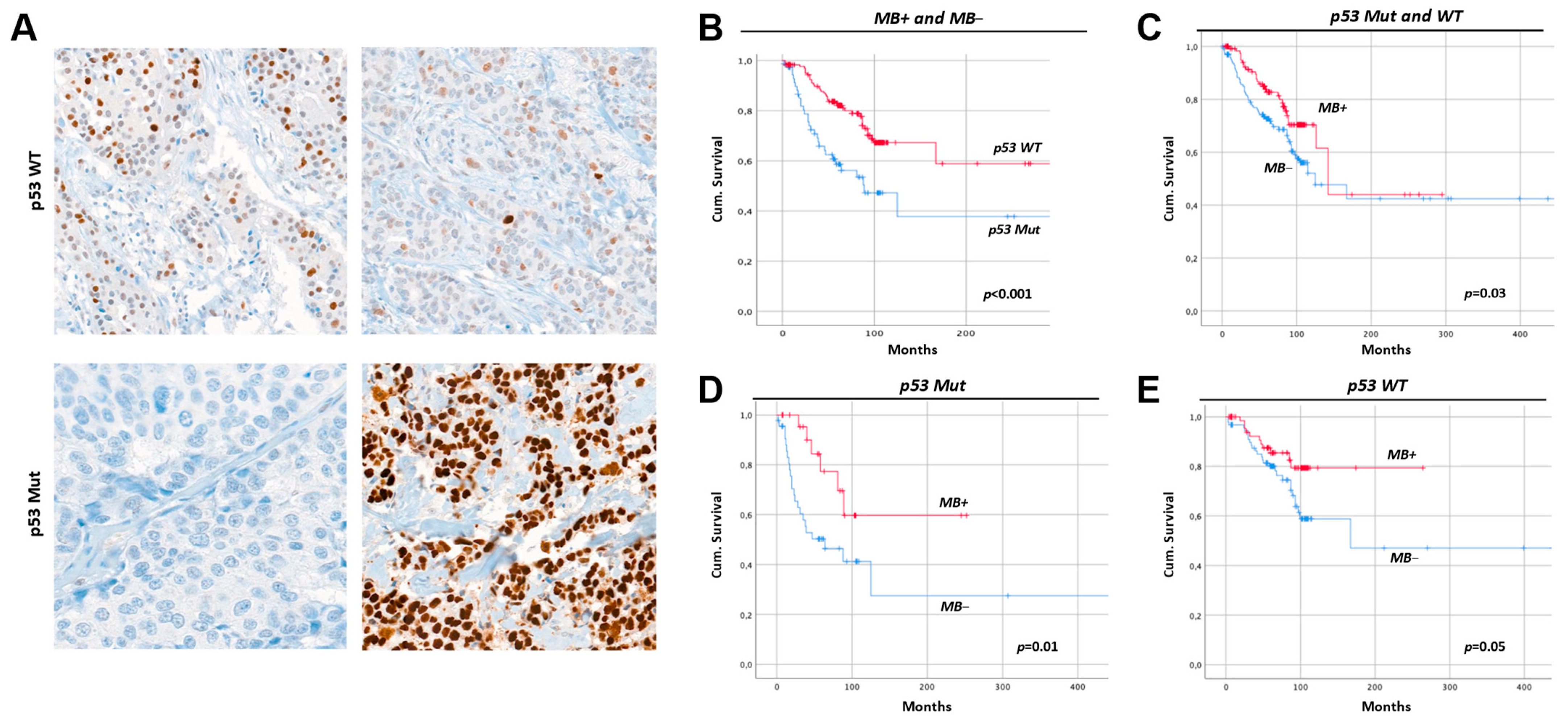
2.8. Breast Cancers with Mutant p53 Have Less MB and a Worse Prognosis
3. Discussion
3.1. Myoglobin Attenuates Normoxic Breast Cancer Cell Survival and ERα Signaling While Enhancing Apoptosis and Response to Chemotherapy by Increasing ROS Levels
3.2. Myoglobin Regulates the Migratory Capacity of Hypoxic Cancer Cells
3.3. Myoglobin in MCF7 vs. SKBR3 and the Hypothesized Interplay with p53
4. Materials and Methods
4.1. Cancer Cells Lines and Cell Culture
4.2. Generation of CRISPR/Cas9-Mediated Knockouts of MB
4.2.1. Preparation of CRISPR Plasmids against Human MB
4.2.2. Generation of MB Knockout Clones of MCF7 and SKBR3 Cells
4.3. Total Protein Extraction and Western Blotting
4.4. RNA Extraction and Real-Time PCR
4.5. Trypan Blue Viability Assay
4.6. Colony Forming Assay
4.7. BrdU Proliferation Assay
4.8. MTT Assay
4.9. Migration Assay
4.10. Cell Cycle Status Analysis
4.11. Annexin-V and NAO-Based FACS Analysis of Apoptosis
4.12. Invasion Assay
4.13. ROS Measurement
4.14. Immunofluorescence
4.15. Myoglobin Expression
4.16. RNA Sequencing
4.16.1. RNA Quality Control and Sequencing
4.16.2. Mapping against the Reference Genome
4.16.3. Gene Ontology Analyses
4.17. Human Cancer Tissue Array Staining
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gödecke, A.; Flögel, U.; Zanger, K.; Ding, Z.; Hirchenhain, J.; Decking, U.K.M.; Schrader, J. Disruption of myoglobin in mice induces multiple compensatory mechanisms. Proc. Natl. Acad. Sci. USA 1999, 96, 10495–10500. [Google Scholar] [CrossRef] [PubMed]
- Endeward, V.; Gros, G.; Jürgens, K.D. Significance of myoglobin as an oxygen store and oxygen transporter in the intermittently perfused human heart: A model study. Cardiovasc. Res. 2010, 87, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Grange, R.W.; Meeson, A.; Chin, E.; Lau, K.S.; Stull, J.T.; Shelton, J.M.; Williams, R.S.; Garry, D.J. Functional and molecular adaptations in skeletal muscle of myoglobin-mutant mice. Am. J. Physiol. Physiol. 2001, 281, C1487–C1494. [Google Scholar] [CrossRef] [PubMed]
- Flögel, U.; Gödecke, A.; Klotz, L.; Schrader, J. Role of myoglobin in the antioxidant defense of the heart. FASEB J. 2004, 18, 1156–1158. [Google Scholar] [CrossRef]
- Flögel, U.; Merx, M.W.; Gödecke, A.; Decking, U.K.M.; Schrader, J. Myoglobin: A scavenger of bioactive NO. Proc. Natl. Acad. Sci. USA 2001, 98, 735–740. [Google Scholar] [CrossRef]
- Hendgen-Cotta, U.B.; Merx, M.W.; Shiva, S.; Schmitz, J.; Becher, S.; Klare, J.P.; Steinhoff, H.-J.; Goedecke, A.; Schrader, J.; Gladwin, M.T.; et al. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2008, 105, 10256–10261. [Google Scholar] [CrossRef]
- Flonta, S.E.; Arena, S.; Pisacane, A.; Michieli, P.; Bardelli, A. Expression and Functional Regulation of Myoglobin in Epithelial Cancers. Am. J. Pathol. 2009, 175, 201–206. [Google Scholar] [CrossRef]
- Kristiansen, G.; Rose, M.; Geisler, C.; Fritzsche, F.R.; Gerhardt, J.; Lüke, C.; Ladhoff, A.-M.; Knüchel, R.; Dietel, M.; Moch, H.; et al. Endogenous myoglobin in human breast cancer is a hallmark of luminal cancer phenotype. Br. J. Cancer 2010, 102, 1736–1745. [Google Scholar] [CrossRef]
- Meller, S.; Van Ellen, A.; Gevensleben, H.; Bicker, A.; Hankeln, T.; Gorr, T.A.; Sailer, V.; Dröge, F.; Schröck, F.; Bootz, F.; et al. Ectopic Myoglobin Expression Is Associated with a Favourable Outcome in Head and Neck Squamous Cell Carcinoma Patients. Anticancer Res. 2016, 36, 6235–6242. [Google Scholar] [CrossRef]
- Meller, S.; Bicker, A.; Montani, M.; Ikenberg, K.; Rostamzadeh, B.; Sailer, V.; Wild, P.; Dietrich, D.; Uhl, B.; Sulser, T.; et al. Myoglobin expression in prostate cancer is correlated to androgen receptor expression and markers of tumor hypoxia. Virchows Arch. 2014, 465, 419–427. [Google Scholar] [CrossRef]
- Oleksiewicz, U.; Daskoulidou, N.; Liloglou, T.; Tasopoulou, K.; Bryan, J.; Gosney, J.R.; Field, J.K.; Xinarianos, G. Neuroglobin and myoglobin in non-small cell lung cancer: Expression, regulation and prognosis. Lung Cancer 2011, 74, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Ruck, P.; Horny, H.P.; Greschniok, A.; Wehrmann, M.; Kaiserling, E. Nonspecific immunostaining of blast cells of acute leukemia by antibodies against nonhemopoietic antigens. Hematol. Pathol. 1995, 9, 49–56. [Google Scholar] [PubMed]
- Kristiansen, G.; Hu, J.; Wichmann, D.; Stiehl, D.; Rose, M.; Gerhardt, J.; Bohnert, A.; Haaf, A.T.; Moch, H.; Raleigh, J.; et al. Endogenous Myoglobin in Breast Cancer Is Hypoxia-inducible by Alternative Transcription and Functions to Impair Mitochondrial Activity. J. Biol. Chem. 2011, 286, 43417–43428. [Google Scholar] [CrossRef]
- Galluzzo, M.; Pennacchietti, S.; Rosano, S.; Comoglio, P.M.; Michieli, P. Prevention of hypoxia by myoglobin expression in human tumor cells promotes differentiation and inhibits metastasis. J. Clin. Investig. 2009, 119, 865–875. [Google Scholar] [CrossRef]
- Cartoni, A.; Menna, P.; Salvatorelli, E.; Braghiroli, D.; Giampietro, R.; Animati, F.; Urbani, A.; Del Boccio, P.; Minotti, G. Oxidative Degradation of Cardiotoxic Anticancer Anthracyclines to Phthalic Acids. J. Biol. Chem. 2004, 279, 5088–5099. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, A.; Kuperwasser, C. The origin of breast tumor heterogeneity. Oncogene 2015, 34, 5309–5316. [Google Scholar] [CrossRef]
- Chintapalli, S.V.; Bhardwaj, G.; Patel, R.; Shah, N.; Patterson, R.L.; van Rossum, D.B.; Anishkin, A.; Adams, S.H. Molecular Dynamic Simulations Reveal the Structural Determinants of Fatty Acid Binding to Oxy-Myoglobin. PLoS ONE 2015, 10, e0128496. [Google Scholar] [CrossRef]
- Shih, L.; Chung, Y.; Sriram, R.; Jue, T. Palmitate interaction with physiological states of myoglobin. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1840, 656–666. [Google Scholar] [CrossRef]
- Aboouf, M.A.; Armbruster, J.; Thiersch, M.; Gassmann, M.; Gödecke, A.; Gnaiger, E.; Kristiansen, G.; Bicker, A.; Hankeln, T.; Zhu, H.; et al. Myoglobin, expressed in brown adipose tissue of mice, regulates the content and activity of mitochondria and lipid droplets. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2021, 1866, 159026. [Google Scholar] [CrossRef]
- Bicker, A.; Brahmer, A.M.; Meller, S.; Kristiansen, G.; Gorr, T.A.; Hankeln, T. The Distinct Gene Regulatory Network of Myoglobin in Prostate and Breast Cancer. PLoS ONE 2015, 10, e0142662. [Google Scholar] [CrossRef] [Green Version]
- Bicker, A.; Dietrich, D.; Gleixner, E.; Kristiansen, G.; Gorr, T.A.; Hankeln, T. Extensive transcriptional complexity during hypoxia-regulated expression of the myoglobin gene in cancer. Hum. Mol. Genet. 2013, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Bicker, A.; Nauth, T.; Gerst, D.; Aboouf, M.; Fandrey, J.; Kristiansen, G.; Gorr, T.A.; Hankeln, T. The role of myoglobin in epithelial cancers: Insights from transcriptomics. Int. J. Mol. Med. 2019, 45, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Gorr, T.A. Hypometabolism as the ultimate defence in stress response: How the comparative approach helps understanding of medically relevant questions. Acta Physiol. 2016, 219, 409–440. [Google Scholar] [CrossRef]
- Fluck, M.M.; Schaffhausen, B.S. Lessons in Signaling and Tumorigenesis from Polyomavirus Middle T Antigen. Microbiol. Mol. Biol. Rev. 2009, 73, 542–563. [Google Scholar] [CrossRef]
- Lima, J.F.; Nofech-Mozes, S.; Bayani, J.; Bartlett, J.M.S. EMT in Breast Carcinoma—A Review. J. Clin. Med. 2016, 5, 65. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Feola, A.; Zuchegna, C.; Romano, A.; Donini, C.F.; Bartollino, S.; Costagliola, C.; Frunzio, R.; Laccetti, P.; Di Domenico, M.; et al. The p85 Regulatory Subunit of PI3K Mediates cAMP-PKA and Insulin Biological Effects on MCF-7 Cell Growth and Motility. Sci. World J. 2014, 2014, 565839. [Google Scholar] [CrossRef]
- Li, C.-L.; Yang, D.; Cao, X.; Wang, F.; Hong, D.-Y.; Wang, J.; Shen, X.-C.; Chen, Y. Fibronectin induces epithelial-mesenchymal transition in human breast cancer MCF-7 cells via activation of calpain. Oncol. Lett. 2017, 13, 3889–3895. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kang, S.-E.; Ahn, K.S.; Um, J.-Y.; Yang, W.M.; Yun, M.; Lee, S.-G. Inhibition of the PI3K-AKT-mTOR pathway suppresses the adipocyte-mediated proliferation and migration of breast cancer cells. J. Cancer 2020, 11, 2552–2559. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Calligé, M.; Kieffer, I.; Richard-Foy, H. CSN5/Jab1 Is Involved in Ligand-Dependent Degradation of Estrogen Receptor α by the Proteasome. Mol. Cell. Biol. 2005, 25, 4349–4358. [Google Scholar] [CrossRef] [Green Version]
- Valley, C.C.; Métivier, R.; Solodin, N.M.; Fowler, A.M.; Mashek, M.T.; Hill, L.; Alarid, E.T. Differential Regulation of Estrogen-Inducible Proteolysis and Transcription by the Estrogen Receptor α N Terminus. Mol. Cell. Biol. 2005, 25, 5417–5428. [Google Scholar] [CrossRef]
- Yang, Z.; Heater, B.S.; Cuddington, C.T.; Palmer, A.F.; Lee, M.M.; Chan, M.K. Targeted Myoglobin Delivery as a Strategy for Enhancing the Sensitivity of Hypoxic Cancer Cells to Radiation. iScience 2020, 23, 101158. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Funes, H.; Coronado, C. Role of anthracyclines in the era of targeted therapy. Cardiovasc. Toxicol. 2007, 7, 56–60. [Google Scholar] [CrossRef]
- Zhou, J.; Kong, D.; Zhang, X.; Wang, Y.; Feng, Z.; Zhang, X.; Zhang, L.; Wang, Y.; Xie, Y.; Chen, X. Myoglobin-Induced Apoptosis: Two Pathways Related to Endoplasmic Reticulum Stress. Ther. Apher. Dial. 2012, 16, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, G.V. Flow-cytometry assay for apoptosis using fluorophor 10-N-nonyl acridine orange. Biochem. Suppl. Ser. A Membr. Cell Biol. 2010, 4, 349–357. [Google Scholar] [CrossRef]
- Jänicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res. Treat. 2008, 117, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, S.; Gu, J.; Honda, T.; McDonnell, T.J.; Swisher, S.G.; Roth, J.A.; Fang, B. Deficiency of caspase-3 in MCF7 cells blocks Bax-mediated nuclear fragmentation but not cell death. Clin. Cancer Res. 2001, 7, 1474–1480. [Google Scholar] [PubMed]
- Capo-Chichi, C.D.; Cai, K.Q.; Smedberg, J.; Ganjei-Azar, P.; Godwin, A.K.; Xu, X.X. Loss of A-type lamin expression compromises nuclear envelope integrity in breast cancer. Chin. J. Cancer 2011, 30, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Murahashi, H.; Azuma, H.; Zamzami, N.; Furuya, K.-J.; Ikebuchi, K.; Yamaguchi, M.; Yamada, Y.; Sato, N.; Fujihara, M.; Kroemer, G.; et al. Possible contribution of apoptosis-inducing factor (AIF) and reactive oxygen species (ROS) to UVB-induced caspase-independent cell death in the T cell line Jurkat. J. Leukoc. Biol. 2003, 73, 399–406. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.-M.; Liu, Z.-G. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2006, 40, 928–939. [Google Scholar] [CrossRef]
- Braganza, A.; Quesnelle, K.; Bickta, J.; Reyes, C.; Wang, Y.; Jessup, M.; Croix, C.S.; Arlotti, J.; Singh, S.V.; Shiva, S. Myoglobin induces mitochondrial fusion, thereby inhibiting breast cancer cell proliferation. J. Biol. Chem. 2019, 294, 7269–7282. [Google Scholar] [CrossRef]
- Bièche, I.; Olivi, M.; Noguès, C.; Vidaud, M.; Lidereau, R. Prognostic value of CCND1 gene status in sporadic breast tumours, as determined by real-time quantitative PCR assays. Br. J. Cancer 2002, 86, 580–586. [Google Scholar] [CrossRef]
- Inoue, K.; Fry, E.A. Aberrant Expression of Cyclin D1 in Cancer. Signal Transduct. Insights 2015, 4, STI.S30306. [Google Scholar] [CrossRef]
- Weitsman, G.E.; Weebadda, W.; Ung, K.; Murphy, L.C. Reactive oxygen species induce phosphorylation of serine 118 and 167 on estrogen receptor alpha. Breast Cancer Res. Treat. 2008, 118, 269–279. [Google Scholar] [CrossRef]
- Bailey, S.T.; Shin, H.; Westerling, T.; Liu, X.S.; Brown, M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 18060–18065. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Wallace, D.C.; Levin, E.R. Functional Estrogen Receptors in the Mitochondria of Breast Cancer Cells. Mol. Biol. Cell 2006, 17, 2125–2137. [Google Scholar] [CrossRef]
- Skulachev, V. Mitochondria in the Programmed Death Phenomena; A Principle of Biology: "It Is Better to Die than to be Wrong". IUBMB Life 2000, 49, 365–373. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Shoshan-Barmatz, V. Reducing VDAC1 expression induces a non-apoptotic role for pro-apoptotic proteins in cancer cell differentiation. Biochim. Biophys. Acta 2016, 1857, 1228–1242. [Google Scholar] [CrossRef]
- Trindade, D.; Pereira, C.; Chaves, S.R.; Manon, S.; Côrte-Real, M.; Sousa, M.J. VDAC regulates AAC-mediated apoptosis and cytochrome c release in yeast. Microb. Cell 2016, 3, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yin, S.; Luo, B.; Wu, X.; Yan, H.; Yan, D.; Chen, C.; Guan, F.; Yuan, J. VDAC1 Conversely Correlates with Cytc Expression and Predicts Poor Prognosis in Human Breast Cancer Patients. Oxidative Med. Cell. Longev. 2021, 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aboouf, M.A.; Guscetti, F.; von Büren, N.; Armbruster, J.; Ademi, H.; Ruetten, M.; Meléndez-Rodríguez, F.; Rülicke, T.; Seymer, A.; Jacobs, R.A.; et al. Erythropoietin receptor regulates tumor mitochondrial biogenesis through iNOS and pAKT. Front. Oncol. 2022, 12, 976961. [Google Scholar] [CrossRef] [PubMed]
- Lüönd, F.; Sugiyama, N.; Bill, R.; Bornes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 2021, 56, 3203–3221.e11. [Google Scholar] [CrossRef] [PubMed]
- Mota, A.D.L.; Evangelista, A.F.; Macedo, T.; Oliveira, R.; Scapulatempo-Neto, C.; Vieira, R.A.; Marques, M.M.C. Molecular characterization of breast cancer cell lines by clinical immunohistochemical markers. Oncol. Lett. 2017, 13, 4708–4712. [Google Scholar] [CrossRef]
- Runnebaum, I.B.; Nagarajan, M.; Bowman, M.; Soto, D.; Sukumar, S. Mutations in p53 as potential molecular markers for human breast cancer. Proc. Natl. Acad. Sci. USA 1991, 88, 10657–10661. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Choy, M.-K.; Movassagh, M.; Siggens, L.; Vujic, A.; Goddard, M.; Sanchez, A.M.; Perkins, N.; Figg, N.; Bennett, M.; Carroll, J.; et al. High-throughput sequencing identifies STAT3 as the DNA-associated factor for p53-NF-kappaB-complex-dependent gene expression in human heart failure. Genome Med. 2010, 2, 37. [Google Scholar] [CrossRef]
- Sermeus, A.; Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, e164. [Google Scholar] [CrossRef]
- Totzeck, M.; Hendgen-Cotta, U.B.; Kelm, M.; Rassaf, T. Crosstalk between Nitrite, Myoglobin and Reactive Oxygen Species to Regulate Vasodilation under Hypoxia. PLoS ONE 2014, 9, e105951. [Google Scholar] [CrossRef]
- Cong, L.; Zhang, F. Genome Engineering Using CRISPR-Cas9 System. In Springer Protocols Handbooks; Springer: Singapore, 2015; Volume 1239, pp. 197–217. [Google Scholar]
- Olson, J.; Mathews, A.J.; Rohlfs, R.J.; Springer, B.A.; Egeberg, K.D.; Sligar, S.G.; Tame, J.; Renaud, J.-P.; Nagai, K. The role of the distal histidine in myoglobin and haemoglobin. Nature 1988, 336, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Kim, K.H.; Sederstrom, J.M. Assaying Cell Cycle Status Using Flow Cytometry. Curr. Protoc. Mol. Biol. 2015, 111, 28.6.1–28.6.11. [Google Scholar] [CrossRef]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef]
- Li, J.; Guo, Y.; Duan, L.; Hu, X.; Zhang, X.; Hu, J.; Huang, L.; He, R.; Hu, Z.; Luo, W.; et al. AKR1B10 promotes breast cancer cell migration and invasion via activation of ERK signaling. Oncotarget 2017, 8, 33694–33703. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p53 Status | MB Expression n (%) | p | ||||
|---|---|---|---|---|---|---|
| No | Mild | Moderate | Strong | Total | ||
| Mutant | 28 (32.9) | 26 (30.6) | 23 (27.1) | 8 (9.4) | 85 (100) | 0.017 |
| Wildtype | 39 (19.2) | 66 (32.5) | 70 (34.5) | 28 (13.8) | 203 (100) | |
| Total | 67 (23.3) | 92 (31.9) | 93 (32.3) | 36 (12.5) | 288 (100) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboouf, M.A.; Armbruster, J.; Thiersch, M.; Guscetti, F.; Kristiansen, G.; Schraml, P.; Bicker, A.; Petry, R.; Hankeln, T.; Gassmann, M.; et al. Pro-Apoptotic and Anti-Invasive Properties Underscore the Tumor-Suppressing Impact of Myoglobin on a Subset of Human Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 11483. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911483
Aboouf MA, Armbruster J, Thiersch M, Guscetti F, Kristiansen G, Schraml P, Bicker A, Petry R, Hankeln T, Gassmann M, et al. Pro-Apoptotic and Anti-Invasive Properties Underscore the Tumor-Suppressing Impact of Myoglobin on a Subset of Human Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(19):11483. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911483
Chicago/Turabian StyleAboouf, Mostafa A., Julia Armbruster, Markus Thiersch, Franco Guscetti, Glen Kristiansen, Peter Schraml, Anne Bicker, Ruben Petry, Thomas Hankeln, Max Gassmann, and et al. 2022. "Pro-Apoptotic and Anti-Invasive Properties Underscore the Tumor-Suppressing Impact of Myoglobin on a Subset of Human Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 19: 11483. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911483