
Proteomics Profiling of Stool Samples from Preterm Neonates with SWATH/DIA Mass Spectrometry for Predicting Necrotizing Enterocolitis
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
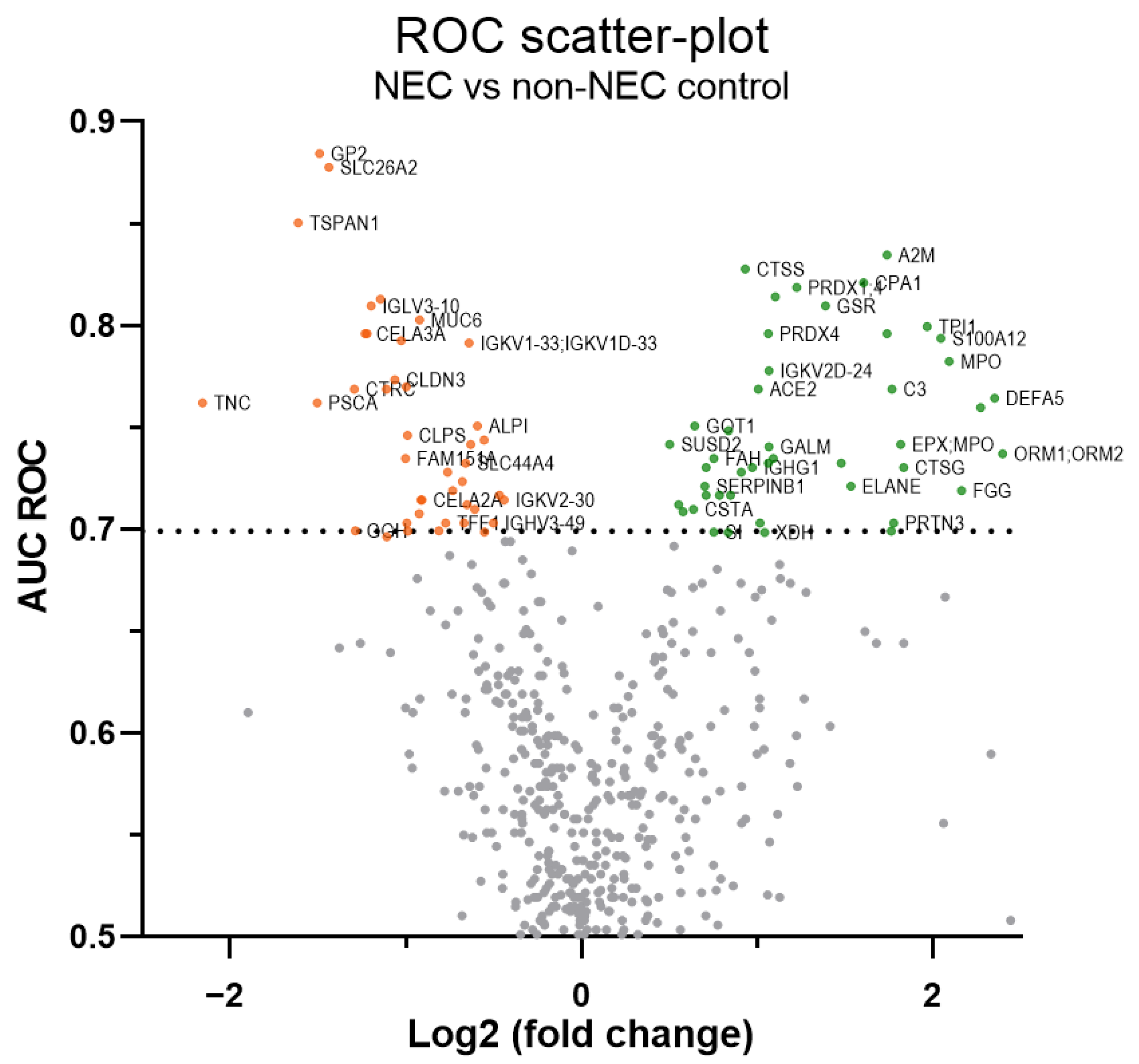
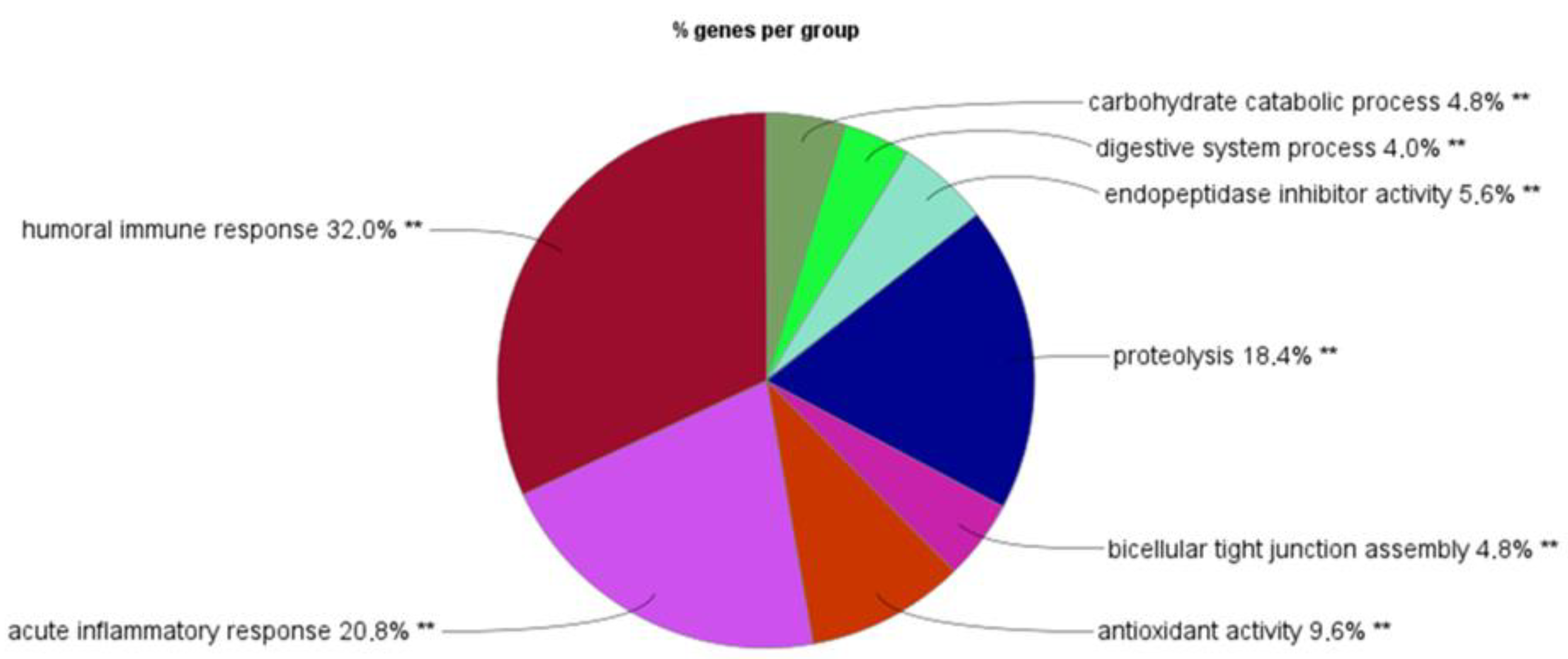
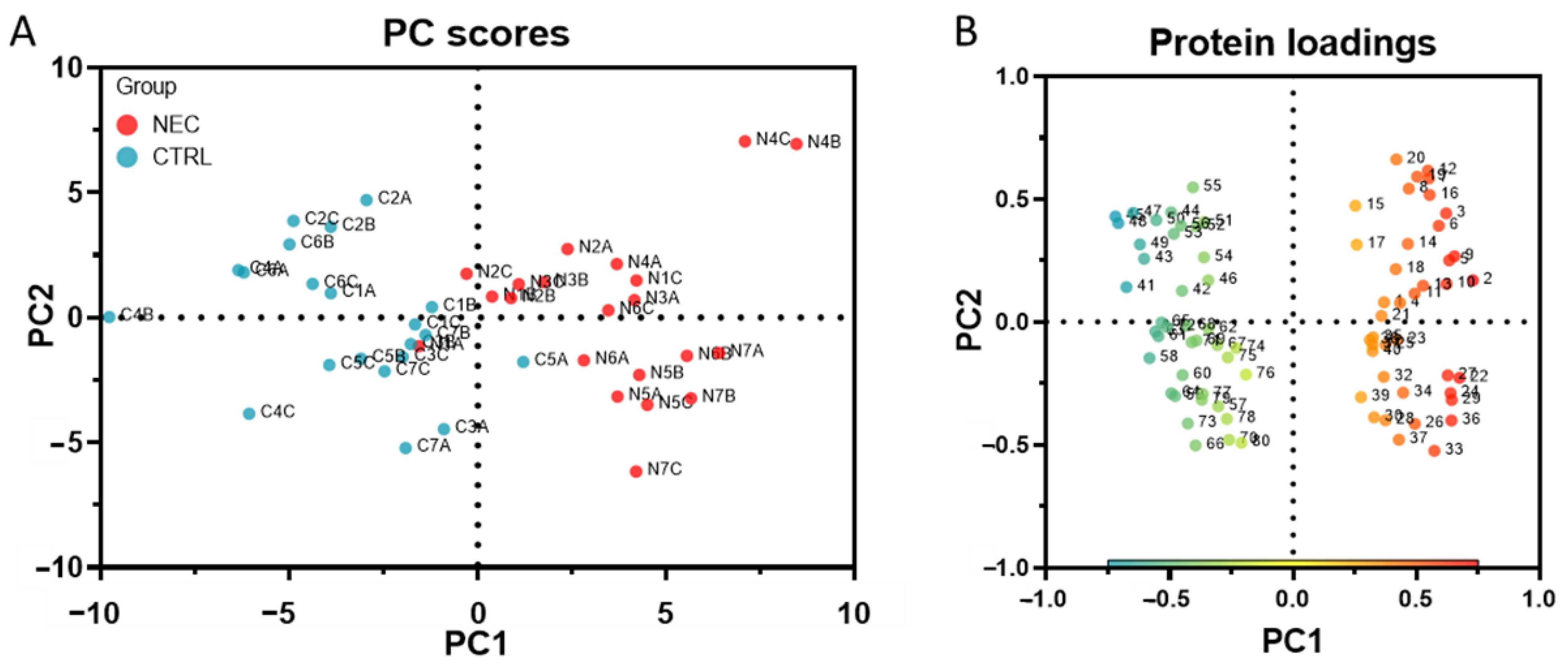
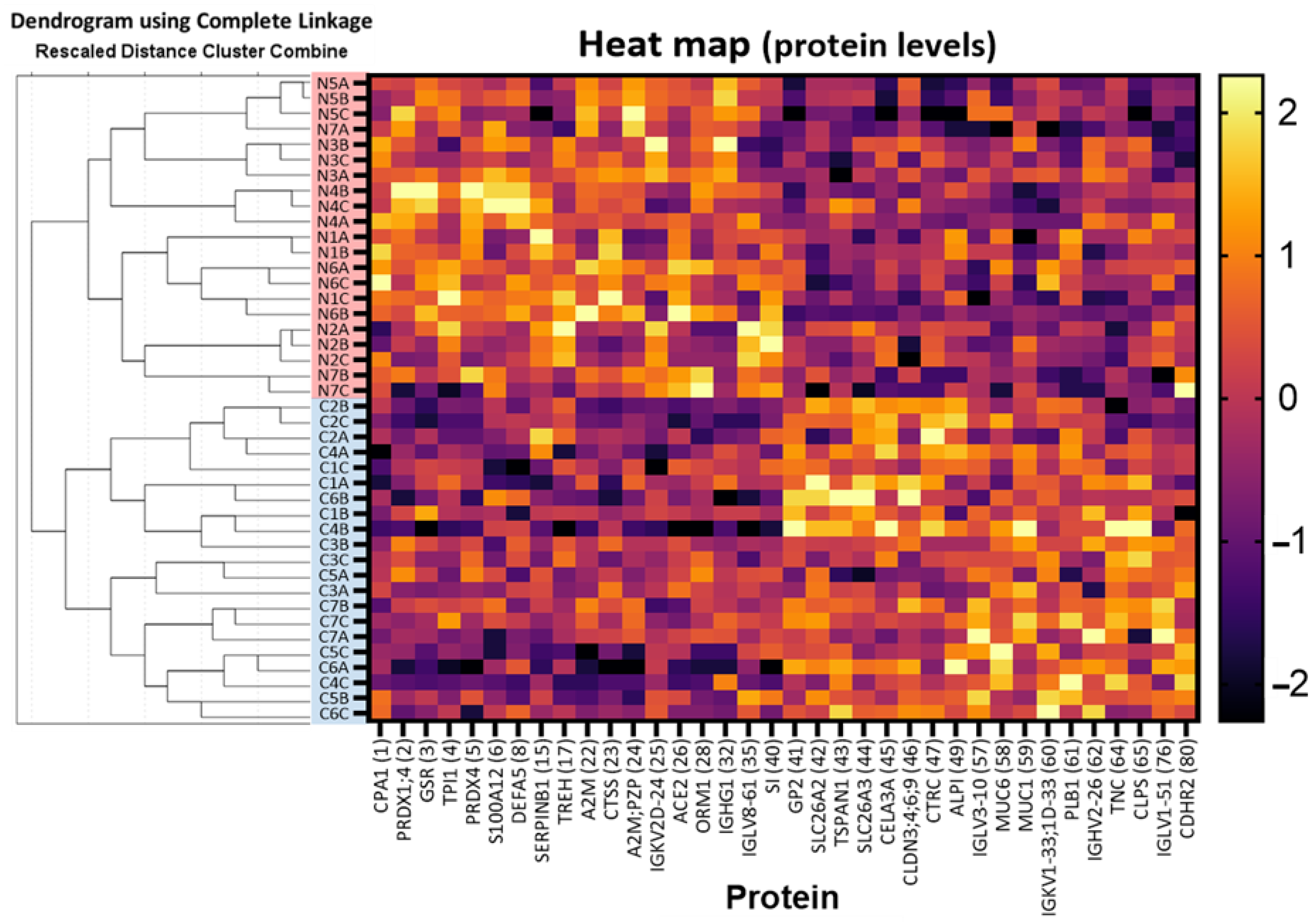
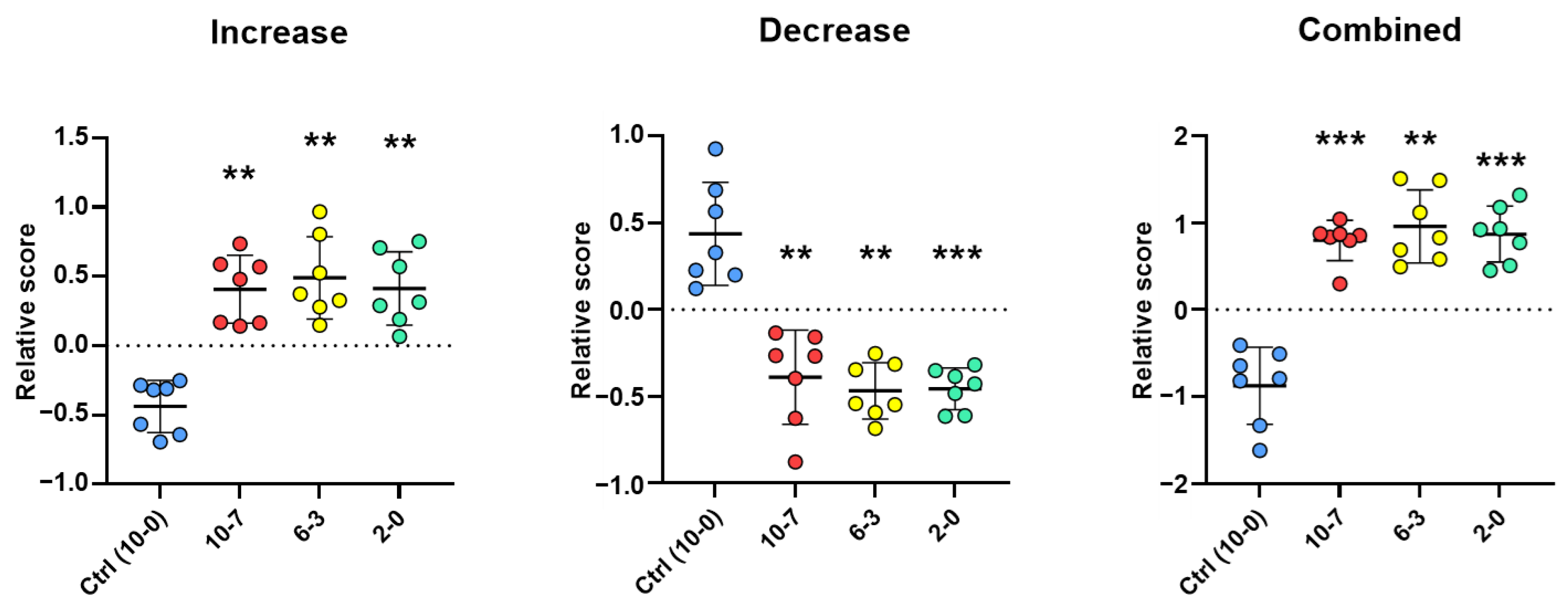
2.1. Discovery of Up- and Down-Regulated Proteins in Stools of NEC Infants by SWATH-MS Analysis
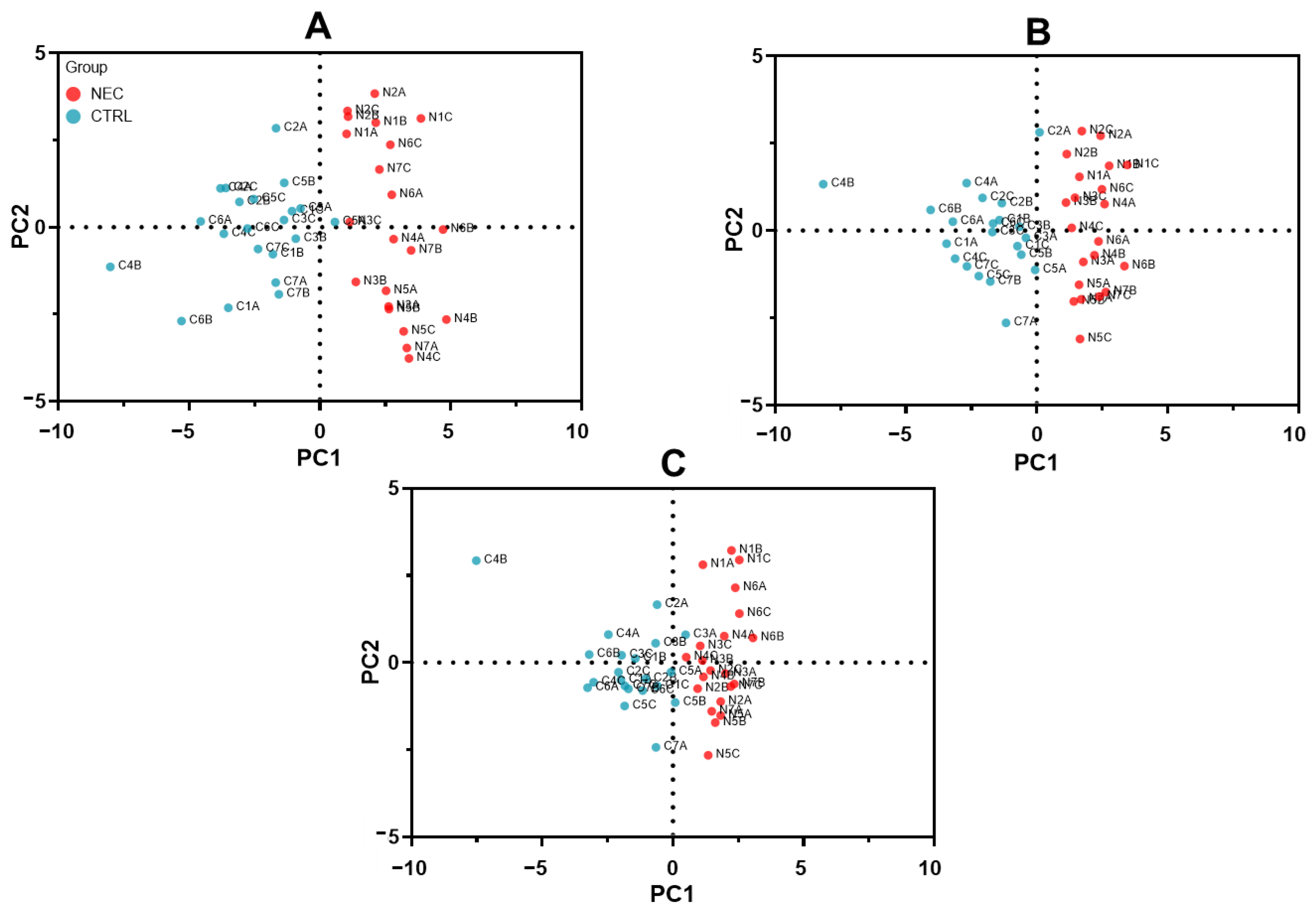
2.2. Optimization of SWATH-MS-Derived Stool Signatures for NEC Infants
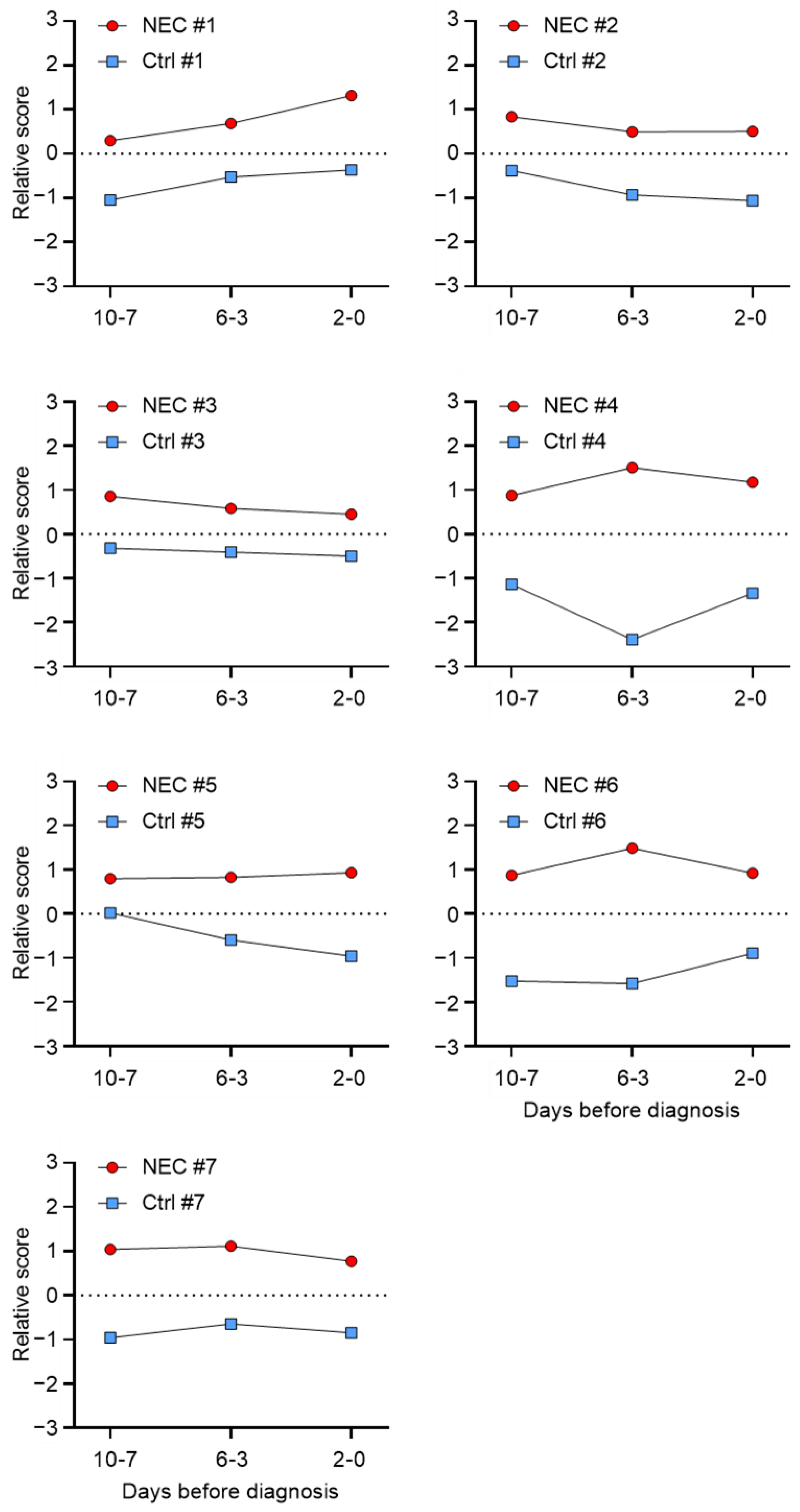
2.3. SWATH-MS-Derived Stool Signature for Predicting NEC Development
3. Discussion
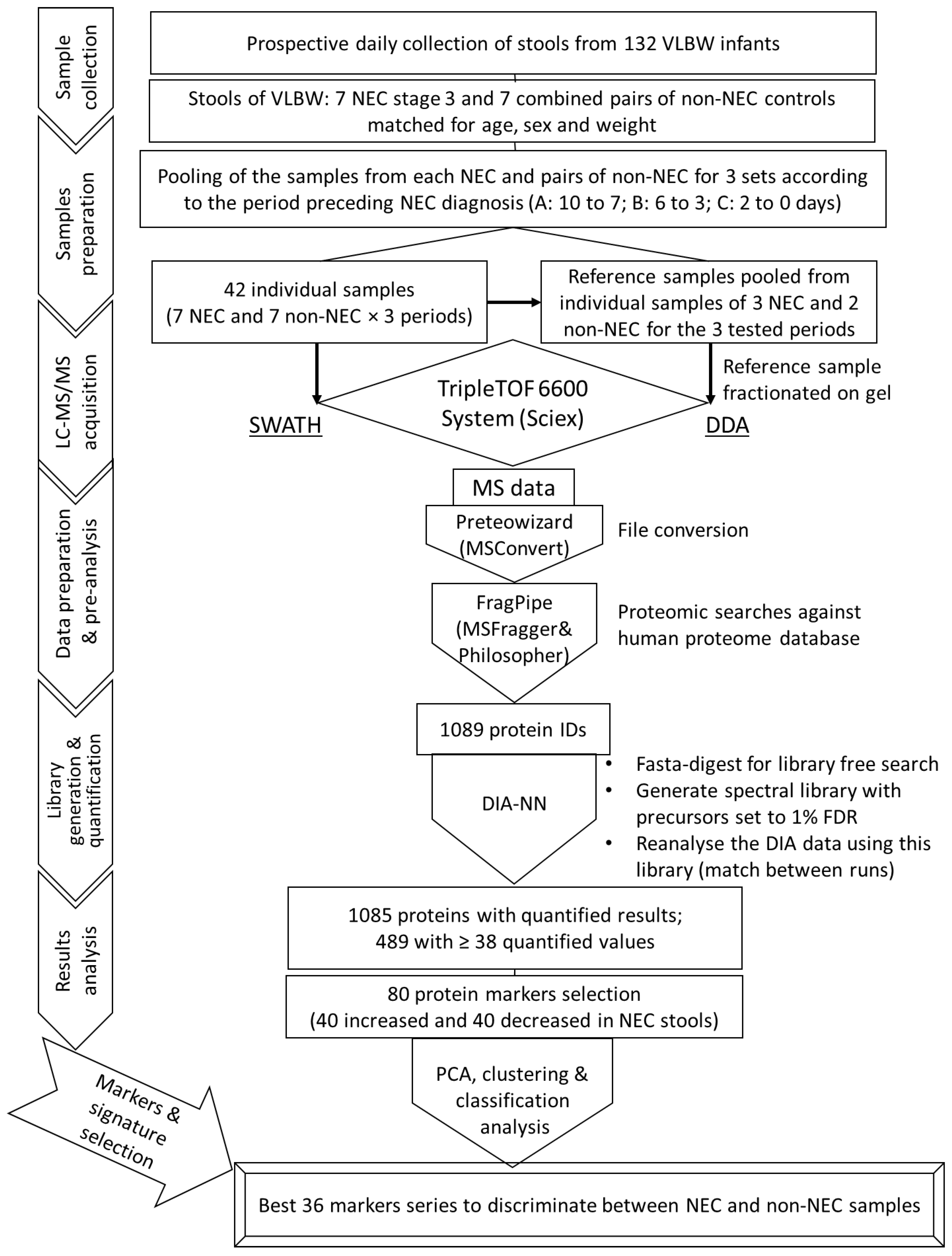
4. Materials and Methods
4.1. Sample Collection and Preparation for LC-MS/MS Analysis
4.2. LC-MS/MS Acquisition
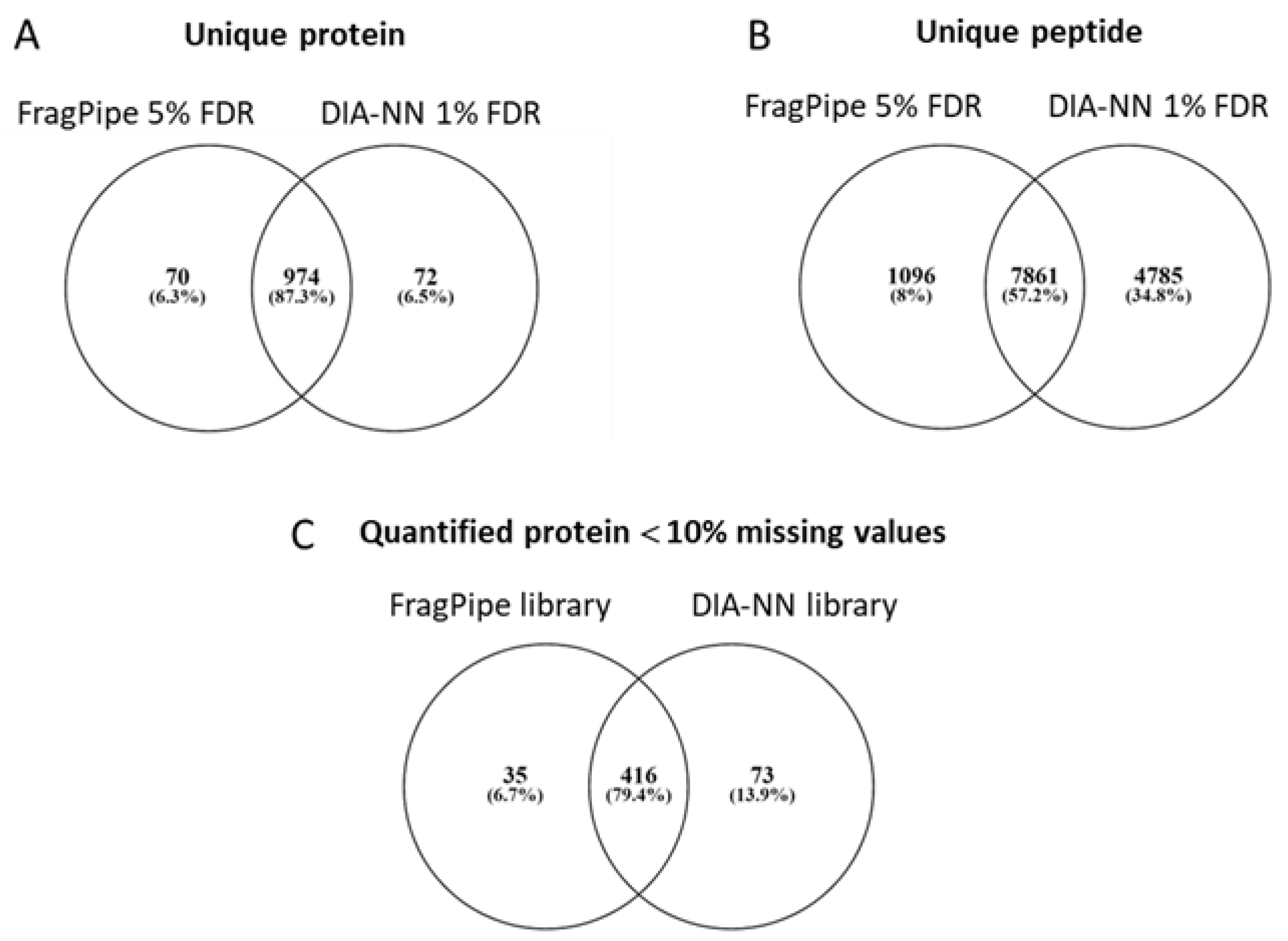
4.3. MS Data Preparation and Pre-Analysis
4.4. Reference Spectral Library Building and Label-Free Quantification Analysis
4.5. Dataset Composition, Protein Marker Selection and Series Evaluation
4.6. Statistical Analysis and Graph
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neu, J.; Walker, W.A. Necrotizing enterocolitis. N. Engl. J. Med. 2011, 364, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, M.C.; Moss, R.L. Necrotizing enterocolitis. Annu. Rev. Med. 2009, 60, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heida, F.H.; Stolwijk, L.; Loos, M.H.; van den Ende, S.J.; Onland, W.; van den Dungen, F.A.; Kooi, E.M.; Bos, A.F.; Hulscher, J.B.; Bakx, R. Increased incidence of necrotizing enterocolitis in the Netherlands after implementation of the new Dutch guideline for active treatment in extremely preterm infants: Results from three academic referral centers. J. Pediatr. Surg. 2017, 52, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Nino, D.F.; Sodhi, C.P.; Hackam, D.J. Necrotizing enterocolitis: New insights into pathogenesis and mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Fitzgibbons, S.C.; Ching, Y.; Yu, D.; Carpenter, J.; Kenny, M.; Weldon, C.; Lillehei, C.; Valim, C.; Horbar, J.D.; Jaksic, T. Mortality of necrotizing enterocolitis expressed by birth weight categories. J. Pediatr. Surg. 2009, 44, 1072–1075; discussion 1075–1076. [Google Scholar] [CrossRef]
- Thyoka, M.; de Coppi, P.; Eaton, S.; Khoo, K.; Hall, N.J.; Curry, J.; Kiely, E.; Drake, D.; Cross, K.; Pierro, A. Advanced necrotizing enterocolitis part 1: Mortality. Eur. J. Pediatr. Surg. 2012, 22, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Jones, I.H.; Hall, N.J. Contemporary Outcomes for Infants with Necrotizing Enterocolitis-A Systematic Review. J. Pediatr. 2020, 220, 86–92.e83. [Google Scholar] [CrossRef]
- Bazacliu, C.; Neu, J. Necrotizing Enterocolitis: Long Term Complications. Curr. Pediatr. Rev. 2019, 15, 115–124. [Google Scholar] [CrossRef]
- Huda, S.; Chaudhery, S.; Ibrahim, H.; Pramanik, A. Neonatal necrotizing enterocolitis: Clinical challenges, pathophysiology and management. Pathophysiology 2014, 21, 3–12. [Google Scholar] [CrossRef]
- Pike, K.; Brocklehurst, P.; Jones, D.; Kenyon, S.; Salt, A.; Taylor, D.; Marlow, N. Outcomes at 7 years for babies who developed neonatal necrotising enterocolitis: The ORACLE Children Study. Arch. Dis. Child. Fetal Neonatal Ed. 2012, 97, F318–F322. [Google Scholar] [CrossRef]
- Alganabi, M.; Lee, C.; Bindi, E.; Li, B.; Pierro, A. Recent advances in understanding necrotizing enterocolitis. F1000Research 2019, 8, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, S.; Rees, C.M.; Hall, N.J. Current Research on the Epidemiology, Pathogenesis, and Management of Necrotizing Enterocolitis. Neonatology 2017, 111, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Bazacliu, C.; Neu, J. Pathophysiology of Necrotizing Enterocolitis: An Update. Curr. Pediatr. Rev. 2019, 15, 68–87. [Google Scholar] [CrossRef] [PubMed]
- Samuels, N.; van de Graaf, R.A.; de Jonge, R.C.J.; Reiss, I.K.M.; Vermeulen, M.J. Risk factors for necrotizing enterocolitis in neonates: A systematic review of prognostic studies. BMC Pediatr. 2017, 17, 105. [Google Scholar] [CrossRef]
- Berkhout, D.J.C.; Klaassen, P.; Niemarkt, H.J.; de Boode, W.P.; Cossey, V.; van Goudoever, J.B.; Hulzebos, C.V.; Andriessen, P.; van Kaam, A.H.; Kramer, B.W.; et al. Risk Factors for Necrotizing Enterocolitis: A Prospective Multicenter Case-Control Study. Neonatology 2018, 114, 277–284. [Google Scholar] [CrossRef]
- Cho, S.X.; Berger, P.J.; Nold-Petry, C.A.; Nold, M.F. The immunological landscape in necrotising enterocolitis. Expert Rev. Mol. Med. 2016, 18, e12. [Google Scholar] [CrossRef] [Green Version]
- Agakidou, E.; Agakidis, C.; Gika, H.; Sarafidis, K. Emerging Biomarkers for Prediction and Early Diagnosis of Necrotizing Enterocolitis in the Era of Metabolomics and Proteomics. Front. Pediatr. 2020, 8, 602255. [Google Scholar] [CrossRef]
- Bell, M.J.; Ternberg, J.L.; Feigin, R.D.; Keating, J.P.; Marshall, R.; Barton, L.; Brotherton, T. Neonatal necrotizing enterocolitis. Therapeutic decisions based upon clinical staging. Ann. Surg. 1978, 187, 1–7. [Google Scholar] [CrossRef]
- Gilfillan, M.; Bhandari, V. Biomarkers for the diagnosis of neonatal sepsis and necrotizing enterocolitis: Clinical practice guidelines. Early Hum. Dev. 2017, 105, 25–33. [Google Scholar] [CrossRef]
- Gephart, S.M.; Gordon, P.V.; Penn, A.H.; Gregory, K.E.; Swanson, J.R.; Maheshwari, A.; Sylvester, K. Changing the paradigm of defining, detecting, and diagnosing NEC: Perspectives on Bell’s stages and biomarkers for NEC. Semin. Pediatr. Surg. 2018, 27, 3–10. [Google Scholar] [CrossRef]
- Wang, K.; Tao, G.; Sylvester, K.G. Recent Advances in Prevention and Therapies for Clinical or Experimental Necrotizing Enterocolitis. Dig. Dis. Sci. 2019, 64, 3078–3085. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Ma, T.P.; Lam, H.S. The use of laboratory biomarkers for surveillance, diagnosis and prediction of clinical outcomes in neonatal sepsis and necrotising enterocolitis. Arch. Dis. Child. Fetal Neonatal Ed. 2015, 100, F448–F452. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.D.; Sharma, D.; Bansal, A. Biomarkers of necrotizing enterocolitis: A review of literature. J. Matern. Fetal Neonatal Med. 2018, 31, 3051–3064. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.C.; Granger, C.L.; Masi, A.C.; Stewart, C.J. Use of omic technologies in early life gastrointestinal health and disease: From bench to bedside. Expert Rev. Proteom. 2021, 18, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Gephart, S.M.; Newnam, K.M. Closing the Gap between Recommended and Actual Human Milk Use for Fragile Infants: What Will It Take to Overcome Disparities? Clin. Perinatol. 2019, 46, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Tao, G.; Sun, Z.; Sylvester, K.G. Recent Potential Noninvasive Biomarkers in Necrotizing Enterocolitis. Gastroenterol. Res. Pract. 2019, 2019, 8413698. [Google Scholar] [CrossRef]
- Floris, I.; Kraft, J.D.; Altosaar, I. Roles of MicroRNA across Prenatal and Postnatal Periods. Int. J. Mol. Sci. 2016, 17, 1994. [Google Scholar] [CrossRef] [Green Version]
- Alexovic, M.; Lindner, J.R.; Bober, P.; Longuespee, R.; Sabo, J.; Davalieva, K. Human peripheral blood mononuclear cells: A review of recent proteomic applications. Proteomics 2022, 22, e2200026. [Google Scholar] [CrossRef]
- Hoffsten, A.; Markasz, L.; Lilja, H.E.; Olsson, K.W.; Sindelar, R. Early Postnatal Comprehensive Biomarkers Cannot Identify Extremely Preterm Infants at Risk of Developing Necrotizing Enterocolitis. Front. Pediatr. 2021, 9, 755437. [Google Scholar] [CrossRef]
- Gollin, G.; Stadie, D.; Mayhew, J.; Slater, L.; Asmerom, Y.; Boskovic, D.; Holden, M.; Angeles, D. Early detection of impending necrotizing enterocolitis with urinary intestinal fatty acid-binding protein. Neonatology 2014, 106, 195–200. [Google Scholar] [CrossRef]
- Coufal, S.; Kokesova, A.; Tlaskalova-Hogenova, H.; Frybova, B.; Snajdauf, J.; Rygl, M.; Kverka, M. Urinary I-FABP, L-FABP, TFF-3, and SAA Can Diagnose and Predict the Disease Course in Necrotizing Enterocolitis at the Early Stage of Disease. J. Immunol. Res. 2020, 2020, 3074313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooven, T.A.; Lin, A.Y.C.; Salleb-Aouissi, A. Multiple Instance Learning for Predicting Necrotizing Enterocolitis in Premature Infants Using Microbiome Data. In Proceedings of the ACM CHIL ’20: ACM Conference on Health, Inference, and Learning, Toronto, ON, Canada, 2–4 April 2020; pp. 99–109. [Google Scholar] [CrossRef] [Green Version]
- Klerk, D.H.; Plosch, T.; Verkaik-Schakel, R.N.; Hulscher, J.B.F.; Kooi, E.M.W.; Bos, A.F. DNA Methylation of TLR4, VEGFA, and DEFA5 Is Associated With Necrotizing Enterocolitis in Preterm Infants. Front. Pediatr. 2021, 9, 630817. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Kehring, A.; Stepp, J.; Calton, C.M.; Gephart, S.M.; Bandlamuri, S.; Boyle, K.E.; Dietz, G.I.; Johnson, H.; Romo, R.E.; et al. Elevated Coefficient of Variation in Total Fecal Bile Acids Precedes Diagnosis of Necrotizing Enterocolitis. Sci. Rep. 2020, 10, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, M.P.; Tremblay, E.; Horth, C.; Fournier-Morin, A.; Grynspan, D.; Babakissa, C.; Levy, E.; Ferretti, E.; Bertelle, V.; Beaulieu, J.F. Lipocalin-2 and calprotectin as stool biomarkers for predicting necrotizing enterocolitis in premature neonates. Pediatr. Res. 2022, 91, 129–136. [Google Scholar] [CrossRef]
- Pergialiotis, V.; Konstantopoulos, P.; Karampetsou, N.; Koutaki, D.; Gkioka, E.; Perrea, D.N.; Papantoniou, N. Calprotectin levels in necrotizing enterocolitis: A systematic review of the literature. Inflamm. Res. 2016, 65, 847–852. [Google Scholar] [CrossRef]
- Salzman, N.H.; Polin, R.A.; Harris, M.C.; Ruchelli, E.; Hebra, A.; Zirin-Butler, S.; Jawad, A.; Martin Porter, E.; Bevins, C.L. Enteric defensin expression in necrotizing enterocolitis. Pediatr. Res. 1998, 44, 20–26. [Google Scholar] [CrossRef] [Green Version]
- MohanKumar, K.; Namachivayam, K.; Song, T.; Jake Cha, B.; Slate, A.; Hendrickson, J.E.; Pan, H.; Wickline, S.A.; Oh, J.Y.; Patel, R.P.; et al. A murine neonatal model of necrotizing enterocolitis caused by anemia and red blood cell transfusions. Nat. Commun. 2019, 10, 3494. [Google Scholar] [CrossRef] [Green Version]
- Heath, M.; Buckley, R.; Gerber, Z.; Davis, P.; Linneman, L.; Gong, Q.; Barkemeyer, B.; Fang, Z.; Good, M.; Penn, D.; et al. Association of Intestinal Alkaline Phosphatase with Necrotizing Enterocolitis Among Premature Infants. JAMA Netw. Open 2019, 2, e1914996. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, E.; Thibault, M.P.; Ferretti, E.; Babakissa, C.; Bertelle, V.; Bettolli, M.; Burghardt, K.M.; Colombani, J.F.; Grynspan, D.; Levy, E.; et al. Gene expression profiling in necrotizing enterocolitis reveals pathways common to those reported in Crohn’s disease. BMC Med. Genom. 2016, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Lualdi, M.; Fasano, M. Statistical analysis of proteomics data: A review on feature selection. J. Proteom. 2019, 198, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, Y.; Luo, Y.; Fu, J.; Zhang, Y.; Li, Y.; Xiao, Z.; Lou, Y.; Qiu, Y.; Zhu, F. Computational advances of tumor marker selection and sample classification in cancer proteomics. Comput. Struct. Biotechnol. J. 2020, 18, 2012–2025. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.M.; Saly, V.; Trivedi, A.; Lesperance, M.L.; Votova, K.; Jackson, A.M.; Croteau, N.S.; Balshaw, R.F.; Bibok, M.B.; Smith, D.S.; et al. Differential Proteomics for Distinguishing Ischemic Stroke from Controls: A Pilot Study of the SpecTRA Project. Transl. Stroke Res. 2018, 9, 590–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, S.B.; Gibson, D.J.; Robertson, P.A.; Pohlmann, J.T.; Fralish, J.S. Parallel Analysis: A method for determining significant principal components. J. Veg. Sci. 1995, 6, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.H. Biostatistics 303. Discriminant analysis. Singap. Med. J. 2005, 46, 54–62. [Google Scholar]
- Neu, J. Necrotizing Enterocolitis: The Future. Neonatology 2020, 117, 240–244. [Google Scholar] [CrossRef]
- Kim, J.H.; Sampath, V.; Canvasser, J. Challenges in diagnosing necrotizing enterocolitis. Pediatr. Res. 2020, 88, 16–20. [Google Scholar] [CrossRef]
- Ng, P.C.; Chan, K.Y.Y.; Lam, H.S.; Wong, R.P.O.; Ma, T.P.Y.; Sit, T.; Leung, K.T.; Chan, L.C.N.; Pang, Y.L.I.; Cheung, H.M.; et al. A Prospective Cohort Study of Fecal miR-223 and miR-451a as Noninvasive and Specific Biomarkers for Diagnosis of Necrotizing Enterocolitis in Preterm Infants. Neonatology 2020, 117, 555–561. [Google Scholar] [CrossRef]
- Lindberg, T.P.; Caimano, M.J.; Hagadorn, J.I.; Bennett, E.M.; Maas, K.; Brownell, E.A.; Matson, A.P. Preterm infant gut microbial patterns related to the development of necrotizing enterocolitis. J. Matern. Fetal Neonatal. Med. 2020, 33, 349–358. [Google Scholar] [CrossRef]
- Mai, V.; Young, C.M.; Ukhanova, M.; Wang, X.; Sun, Y.; Casella, G.; Theriaque, D.; Li, N.; Sharma, R.; Hudak, M.; et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS ONE 2011, 6, e20647. [Google Scholar] [CrossRef]
- Morrow, A.L.; Lagomarcino, A.J.; Schibler, K.R.; Taft, D.H.; Yu, Z.; Wang, B.; Altaye, M.; Wagner, M.; Gevers, D.; Ward, D.V.; et al. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome 2013, 1, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Ang, I.L.; Chiu, R.W.; Li, K.; Lam, H.S.; Wong, R.P.; Chui, K.M.; Cheung, H.M.; Ng, E.W.; Fok, T.F.; et al. Host-response biomarkers for diagnosis of late-onset septicemia and necrotizing enterocolitis in preterm infants. J. Clin. Investig. 2010, 120, 2989–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatziioannou, A.C.; Wolters, J.C.; Sarafidis, K.; Thomaidou, A.; Agakidis, C.; Govorukhina, N.; Kuivenhoven, J.A.; Bischoff, R.; Theodoridis, G. Targeted LC-MS/MS for the evaluation of proteomics biomarkers in the blood of neonates with necrotizing enterocolitis and late-onset sepsis. Anal. Bioanal. Chem. 2018, 410, 7163–7175. [Google Scholar] [CrossRef]
- Murgas Torrazza, R.; Li, N.; Young, C.; Kobeissy, F.; Chow, M.; Chen, S.; Mai, V.; Sharma, R.; Hudak, M.; Shuster, J.; et al. Pilot study using proteomics to identify predictive biomarkers of necrotizing enterocolitis from buccal swabs in very low birth weight infants. Neonatology 2013, 104, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Sylvester, K.G.; Ling, X.B.; Liu, G.Y.; Kastenberg, Z.J.; Ji, J.; Hu, Z.; Wu, S.; Peng, S.; Abdullah, F.; Brandt, M.L.; et al. Urine protein biomarkers for the diagnosis and prognosis of necrotizing enterocolitis in infants. J. Pediatr. 2014, 164, 607–612.e1–7. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Smith, B.; Qvist, N.; Nielsen, C.; Wan, J.M.; Sit, W.H.; Jensen, T.K.; Wang, H.; Sangild, P.T. Intestinal proteome changes during infant necrotizing enterocolitis. Pediatr. Res. 2013, 73, 268–276. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Gotti, C.; Roux-Dalvai, F.; Joly-Beauparlant, C.; Mangnier, L.; Leclercq, M.; Droit, A. Extensive and Accurate Benchmarking of DIA Acquisition Methods and Software Tools Using a Complex Proteomic Standard. J. Proteome Res. 2021, 20, 4801–4814. [Google Scholar] [CrossRef]
- Fernández-Vega, A.; Farabegoli, F.; Alonso-Martínez, M.M.; Ortea, I. Comparison of TIMS-PASEF quantitative proteomics data-analysis workflows using FragPipe, DIA-NN, and Spectronaut from a user’s perspective. bioRxiv 2021. [Google Scholar] [CrossRef]
- Demichev, V.; Yu, F.; Teo, G.C.; Szyrwiel, L.; Rosenberger, G.A.; Decker, J.; Kaspar-Schoenefeld, S.; Lilley, K.S.; Mülleder, M.; Nesvizhskii, A.I.; et al. High sensitivity dia-PASEF proteomics with DIA-NN and FragPipe. bioRxiv 2021. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, E.; Ferretti, E.; Babakissa, C.; Burghardt, K.M.; Levy, E.; Beaulieu, J.F. IL-17-related signature genes linked to human necrotizing enterocolitis. BMC Res. Notes 2021, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, H.J.; Caminero, A.; Turpin, W.; Bermudez-Brito, M.; Santiago, A.; Libertucci, J.; Constante, M.; Raygoza Garay, J.A.; Rueda, G.; Armstrong, S.; et al. Novel Fecal Biomarkers That Precede Clinical Diagnosis of Ulcerative Colitis. Gastroenterology 2021, 160, 1532–1545. [Google Scholar] [CrossRef]
- Lebenthal, E.; Choi, T.S.; Lee, P.C. The development of pancreatic function in premature infants after milk-based and soy-based formulas. Pediatr. Res. 1981, 15, 1240–1244. [Google Scholar] [CrossRef] [Green Version]
- Dabritz, J.; Jenke, A.; Wirth, S.; Foell, D. Fecal phagocyte-specific S100A12 for diagnosing necrotizing enterocolitis. J. Pediatr. 2012, 161, 1059–1064. [Google Scholar] [CrossRef]
- Uchiyama, K.; Naito, Y.; Takagi, T.; Mizushima, K.; Hirai, Y.; Hayashi, N.; Harusato, A.; Inoue, K.; Fukumoto, K.; Yamada, S.; et al. Serpin B1 protects colonic epithelial cell via blockage of neutrophil elastase activity and its expression is enhanced in patients with ulcerative colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1163–G1170. [Google Scholar] [CrossRef]
- Thuijls, G.; Derikx, J.P.; van Wijck, K.; Zimmermann, L.J.; Degraeuwe, P.L.; Mulder, T.L.; Van der Zee, D.C.; Brouwers, H.A.; Verhoeven, B.H.; van Heurn, L.W.; et al. Non-invasive markers for early diagnosis and determination of the severity of necrotizing enterocolitis. Ann. Surg. 2010, 251, 1174–1180. [Google Scholar] [CrossRef]
- Kumar, A.; Priyamvada, S.; Ge, Y.; Jayawardena, D.; Singhal, M.; Anbazhagan, A.N.; Chatterjee, I.; Dayal, A.; Patel, M.; Zadeh, K.; et al. A Novel Role of SLC26A3 in the Maintenance of Intestinal Epithelial Barrier Integrity. Gastroenterology 2021, 160, 1240–1255.e1243. [Google Scholar] [CrossRef]
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 2021, 16, 3737–3760. [Google Scholar] [CrossRef]
- Mann, M.; Kumar, C.; Zeng, W.F.; Strauss, M.T. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef]
- Barkovits, K.; Pacharra, S.; Pfeiffer, K.; Steinbach, S.; Eisenacher, M.; Marcus, K.; Uszkoreit, J. Reproducibility, Specificity and Accuracy of Relative Quantification Using Spectral Library-based Data-independent Acquisition. Mol. Cell. Proteom. 2020, 19, 181–197. [Google Scholar] [CrossRef] [Green Version]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [Green Version]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Xuan, Y.; Sondermann, J.; Schmidt, M.; Gomez-Varela, D.; Reiter, L. Optimization of Experimental Parameters in Data-Independent Mass Spectrometry Significantly Increases Depth and Reproducibility of Results. Mol. Cell. Proteom. MCP 2017, 16, 2296–2309. [Google Scholar] [CrossRef] [Green Version]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef]
- da Veiga Leprevost, F.; Haynes, S.E.; Avtonomov, D.M.; Chang, H.Y.; Shanmugam, A.K.; Mellacheruvu, D.; Kong, A.T.; Nesvizhskii, A.I. Philosopher: A versatile toolkit for shotgun proteomics data analysis. Nat. Methods 2020, 17, 869–870. [Google Scholar] [CrossRef]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Teo, G.C.; Polasky, D.A.; Yu, F.; Nesvizhskii, A.I. Fast Deisotoping Algorithm and Its Implementation in the MSFragger Search Engine. J. Proteome Res. 2021, 20, 498–505. [Google Scholar] [CrossRef]
- Adusumilli, R.; Mallick, P. Data Conversion with ProteoWizard msConvert. Methods Mol. Biol. 2017, 1550, 339–368. [Google Scholar] [CrossRef]
- Tsou, C.C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.C.; Nesvizhskii, A.I. DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.Y.; Kong, A.T.; da Veiga Leprevost, F.; Avtonomov, D.M.; Haynes, S.E.; Nesvizhskii, A.I. Crystal-C: A Computational Tool for Refinement of Open Search Results. J. Proteome Res. 2020, 19, 2511–2515. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Teo, G.C.; Kong, A.T.; Haynes, S.E.; Avtonomov, D.M.; Geiszler, D.J.; Nesvizhskii, A.I. Identification of modified peptides using localization-aware open search. Nat. Commun. 2020, 11, 4065. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. MCP 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; Garcia-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef] [Green Version]
- Meunier, B.; Dumas, E.; Piec, I.; Bechet, D.; Hebraud, M.; Hocquette, J.F. Assessment of hierarchical clustering methodologies for proteomic data mining. J. Proteome Res. 2007, 6, 358–366. [Google Scholar] [CrossRef]
- Steinley, D. K-means clustering: A half-century synthesis. Br. J. Math. Stat. Psychol. 2006, 59, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Huberty, C.J.; Wisenbaker, J.M.; Smith, J.D.; Smith, J.C. Using Categorical Variables in Discriminant Analysis. Multivar. Behav. Res. 1986, 21, 479–496. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 14 March 2022).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Series | 80 Markers | |||
|---|---|---|---|---|
| PC summary | PC1 | PC2 | PC6 | PC41 |
| Eigenvalue | 18.23 | 8.64 | 4.12 | 0.04 |
| Proportion of variance | 22.79% | 10.80% | 5.15% | 0.06% |
| Cumulative proportion of variance | 22.79% | 33.58% | 62.28% | 100.00% |
| Parallel analysis PC selection | Selected | Selected | Last selected | |
| Series | 36 markers | |||
| PC summary | PC1 | PC2 | PC4 | PC36 |
| Eigenvalue | 9.31 | 4.05 | 2.68 | 0.002 |
| Proportion of variance | 25.86% | 11.24% | 7.45% | 0.00005% |
| Cumulative proportion of variance | 25.86% | 37.10% | 54.08% | 100.00% |
| Parallel analysis PC selection | Selected | Selected | Last selected | |
| Series | 20 markers | |||
| PC summary | PC1 | PC2 | PC20 | |
| Eigenvalue | 6.15 | 2.21 | 0.06 | |
| Proportion of variance | 30.77% | 11.05% | 0.28% | |
| Cumulative proportion of variance | 30.77% | 41.82% | 100.00% | |
| Parallel analysis PC selection | Selected | |||
| Series | 14 markers | |||
| PC summary | PC1 | PC2 | PC14 | |
| Eigenvalue | 4.72 | 1.89 | 0.08 | |
| Proportion of variance | 33.74% | 13.49% | 0.60% | |
| Cumulative proportion of variance | 33.74% | 47.22% | 100.00% | |
| Parallel analysis PC selection | Selected | |||
| PC: principal component | ||||
| Samples | 36 Markers | 20 Markers | 14 Markers | |
|---|---|---|---|---|
| Classification | NEC (N) | 21N | 21N | 21N |
| CTRL (C) | 21C | 21C | 21C | |
| Misclassified by period | Gr. A | 0 | 0 | 0 |
| Gr. B | 0 | 0 | 0 | |
| Gr. C | 0 | 0 | 0 |
| Hierarchical Clustering | 36 Markers | 20 Markers | 14 Markers | |
|---|---|---|---|---|
| NEC (N) | Cluster 1 | 21N | 21N | 21N/4C |
| Ctrl (C) | Cluster 2 | 21C | 21C | 17C |
| Misclustered by period | Gr. A | 0 | 0 | 3 |
| Gr. B | 0 | 0 | 1 | |
| Gr. C | 0 | 0 | 0 | |
| K-means clustering | 36 markers | 20 markers | 14 markers | |
| NEC (N) | Cluster 1 | 21N/1C | 21N/2C | 21N/5C |
| Ctrl (C) | Cluster 2 | 20C | 19C | 16C |
| Misclustered by period | Gr. A | 1 | 2 | 4 |
| Gr. B | 0 | 0 | 1 | |
| Gr. C | 0 | 0 | 0 | |
| Linear discriminant analysis | 36 markers | 20 markers | 14 markers | |
| Classified | NEC (N) | 21N | 21N/1C | 21N/1C |
| Ctrl (C) | 21C | 20C | 20C | |
| Wilk’s Lambda | 0.035 | 0.134 | 0.169 | |
| Significance (chi-square) | <0.001 | <0.001 | <0.001 | |
| Misclassified by period | Gr. A | 0 | 1 | 1 |
| Gr. B | 0 | 0 | 0 | |
| Gr. C | 0 | 0 | 0 | |
| No. | First Protein Name 1 | Protein & Protein Groups (Gene Symbol) | Quantified Values (42 Samples) | Fold Change Ratio 2 NEC/Control | ROC Curve AUC | Mann–Whitney Test 3 | |
|---|---|---|---|---|---|---|---|
| p-Value | q-Value | ||||||
| More abundant in NEC stools | |||||||
| 1 | Carboxypeptidase A1 | CPA1 | 42 | 3.04 | 0.82 | <0.001 | 0.003 |
| 2 | Peroxiredoxin-1 | PRDX1;PRDX4 | 39 | 2.33 | 0.82 | <0.001 | 0.003 |
| 3 | Glutathione reductase, mitochondrial | GSR | 42 | 2.61 | 0.81 | <0.001 | 0.003 |
| 4 | Triosephosphate isomerase | TPI1 | 40 | 3.90 | 0.80 | 0.001 | 0.004 |
| 5 | Peroxiredoxin-4 | PRDX4 | 41 | 2.09 | 0.80 | 0.001 | 0.004 |
| 6 | Protein S100-A12 | S100A12 | 39 | 4.12 | 0.79 | 0.001 | 0.004 |
| 8 | Defensin-5 | DEFA5 | 42 | 5.09 | 0.76 | 0.003 | 0.009 |
| 15 | Leukocyte elastase inhibitor | SERPINB1 | 42 | 1.62 | 0.72 | 0.014 | 0.023 |
| 17 | Trehalase | TREH | 41 | 1.63 | 0.72 | 0.016 | 0.023 |
| 22 | Alpha-2-macroglobulin (1) | A2M | 42 | 3.33 | 0.83 | <0.001 | 0.002 |
| 23 | Cathepsin S | CTSS | 42 | 1.91 | 0.83 | <0.001 | 0.003 |
| 24 | Alpha-2-macroglobulin (2) | A2M;PZP | 42 | 3.33 | 0.80 | 0.001 | 0.004 |
| 25 | Probable non-functional Ig kappa variable 2D-24 | IGKV2D-24 | 42 | 2.09 | 0.78 | 0.002 | 0.007 |
| 26 | Angiotensin-converting enzyme 2 | ACE2 | 42 | 2.01 | 0.77 | 0.002 | 0.007 |
| 28 | Alpha-1-acid glycoprotein 1 (1) | ORM1 | 42 | 4.82 | 0.76 | 0.003 | 0.009 |
| 32 | Ig heavy constant gamma 1 | IGHG1 | 42 | 1.96 | 0.73 | 0.010 | 0.018 |
| 35 | Ig lambda variable 8-61 | IGLV8-61 | 42 | 1.80 | 0.72 | 0.016 | 0.023 |
| 40 | Sucrase-isomaltase, intestinal | SI | 42 | 1.68 | 0.70 | 0.052 | 0.055 |
| Less abundant in NEC stools | |||||||
| 41 | Pancreatic secretory granule mb. major gp GP2 | GP2 | 42 | 0.36 | 0.88 | <0.001 | <0.001 |
| 42 | Sulfate transporter | SLC26A2 | 41 | 0.37 | 0.88 | <0.001 | <0.001 |
| 43 | Tetraspanin-1 | TSPAN1 | 42 | 0.33 | 0.85 | <0.001 | 0.001 |
| 44 | Chloride anion exchanger | SLC26A3 | 40 | 0.45 | 0.81 | <0.001 | 0.003 |
| 45 | Chymotrypsin-like elastase family member 3A (1) | CELA3A | 42 | 0.43 | 0.80 | 0.001 | 0.004 |
| 46 | Claudin-3 | CLDN3,4,6,9 | 42 | 0.48 | 0.77 | 0.002 | 0.007 |
| 47 | Chymotrypsin-C | CTRC | 42 | 0.41 | 0.77 | 0.002 | 0.007 |
| 49 | Intestinal-type alkaline phosphatase | ALPI | 42 | 0.66 | 0.75 | 0.005 | 0.012 |
| 57 | Ig lambda variable 3-10 | IGLV3-10 | 42 | 0.44 | 0.81 | <0.001 | 0.003 |
| 58 | Mucin-6 | MUC6 | 42 | 0.53 | 0.80 | 0.001 | 0.004 |
| 59 | Mucin-1 | MUC1 | 42 | 0.43 | 0.80 | 0.001 | 0.004 |
| 60 | Ig kappa variable 1D-33 | IGKV1-33 | 42 | 0.64 | 0.79 | 0.001 | 0.004 |
| 61 | Phospholipase B1, membrane-associated | PLB1 | 38 | 0.49 | 0.79 | 0.001 | 0.004 |
| 62 | Ig heavy variable 2-26 | IGHV2-26 | 38 | 0.50 | 0.77 | 0.002 | 0.007 |
| 64 | Tenascin | TNC | 41 | 0.22 | 0.76 | 0.003 | 0.009 |
| 65 | Colipase | CLPS | 42 | 0.50 | 0.75 | 0.006 | 0.014 |
| 76 | Ig lambda variable 1-51 | IGLV1-51 | 42 | 0.57 | 0.70 | 0.028 | 0.032 |
| 80 | Cadherin-related family member 2 | CDHR2 | 42 | 0.68 | 0.70 | 0.038 | 0.041 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagné, D.; Shajari, E.; Thibault, M.-P.; Noël, J.-F.; Boisvert, F.-M.; Babakissa, C.; Levy, E.; Gagnon, H.; Brunet, M.A.; Grynspan, D.; et al. Proteomics Profiling of Stool Samples from Preterm Neonates with SWATH/DIA Mass Spectrometry for Predicting Necrotizing Enterocolitis. Int. J. Mol. Sci. 2022, 23, 11601. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911601
Gagné D, Shajari E, Thibault M-P, Noël J-F, Boisvert F-M, Babakissa C, Levy E, Gagnon H, Brunet MA, Grynspan D, et al. Proteomics Profiling of Stool Samples from Preterm Neonates with SWATH/DIA Mass Spectrometry for Predicting Necrotizing Enterocolitis. International Journal of Molecular Sciences. 2022; 23(19):11601. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911601
Chicago/Turabian StyleGagné, David, Elmira Shajari, Marie-Pier Thibault, Jean-François Noël, François-Michel Boisvert, Corentin Babakissa, Emile Levy, Hugo Gagnon, Marie A. Brunet, David Grynspan, and et al. 2022. "Proteomics Profiling of Stool Samples from Preterm Neonates with SWATH/DIA Mass Spectrometry for Predicting Necrotizing Enterocolitis" International Journal of Molecular Sciences 23, no. 19: 11601. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911601