
Metabolic Heterogeneity of Brain Tumor Cells of Proneural and Mesenchymal Origin
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
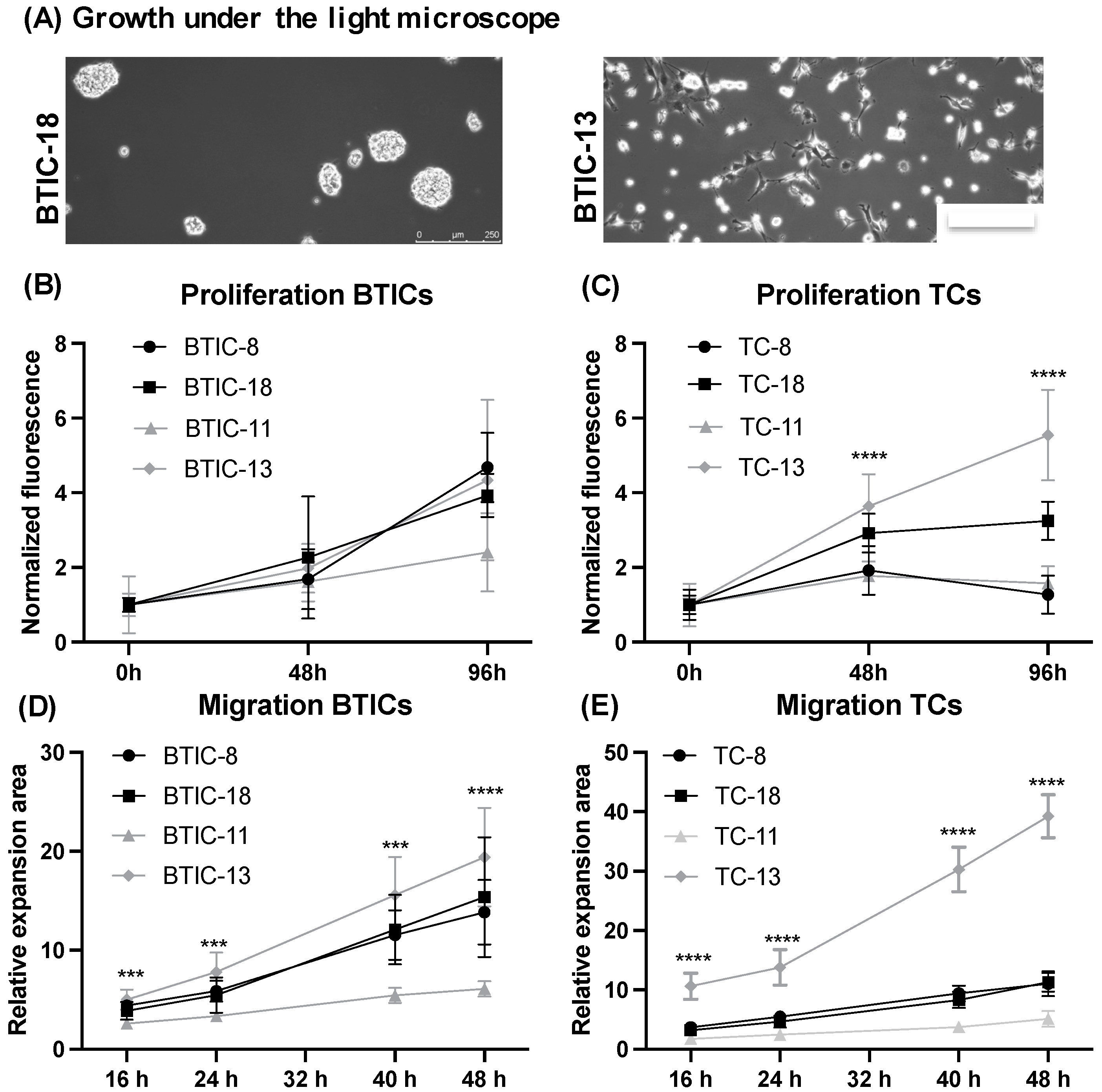
2.1. Characteristics of Proneural and Mesenchymal BTICs and TCs
2.2. Response of Proneural and Mesenchymal BTICs and TCs to OXPHOS Inhibition
2.3. Oxygen Consumption of Proneural and Mesenchymal BTICs and TCs
2.4. Expression of Glycolytic Genes among Proneural and Mesenchymal Cells
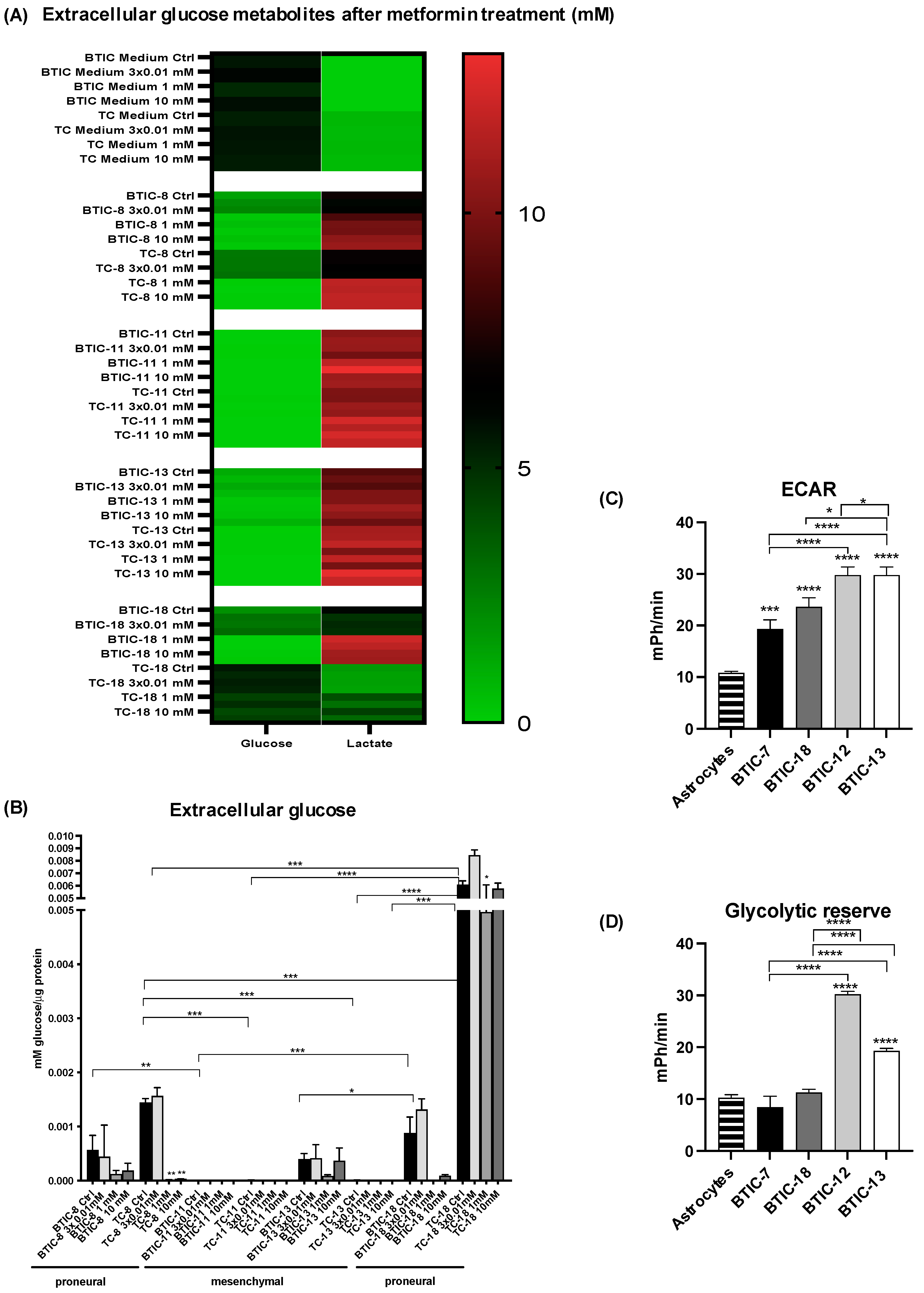
2.5. Activity of Glycolytic Enzymes and Glucose Flux among Proneural and Mesenchymal BTICs
2.6. Relation of Key Findings to Survival Data from TCGA
3. Discussion
4. Materials and Methods
4.1. Cellular Models and Functional Assays
4.1.1. Tumor Specimens and Enrichment of BTICs
4.1.2. Cell Proliferation and Spheroid Migration Assays
4.1.3. Organotypic Brain Slice Cultures (OBSCs)
4.1.4. Treatment of BTICs with Metformin
4.2. RNA and Protein-Based Methods
4.2.1. Polymerase Chain Reaction (PCR) and Quantitative Real Time PCR (qRT-PCR)
4.2.2. Microarray Analysis, Clustering, and Gene Set Enrichment Analysis
4.2.3. RNA Sequencing to Validate Molecular Subgroups from Microarray Analysis
4.2.4. TCGA Analysis
4.2.5. Western Blot
4.3. Metabolic Assays
4.3.1. Enzyme Activity Assays
4.3.2. PreSens Technology
4.3.3. High-Resolution Respirometry
4.3.4. Extracellular Lactate and Glucose Levels
4.3.5. Extracellular Acidification Rate Analysis by Seahorse Technology
4.3.6. 13C-Glucose Isotope Tracing
4.4. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Primers, Antibodies and Supplementary Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qRT-PCR | ||||
|---|---|---|---|---|
| Gene | Forward Primer | Reverse Primer | Source | Annealing Temperature |
| MCT4 | 5′-CAG TTC GAG GTG CTC ATGG-3′ | 5′-ATG TAG ACG TGG GTC GCA TC-3′ | eurofins-mwg/operon | 59 °C |
| HK1 | 5′-GGA CTG GAC CGT CTG AAT GT-3′ | 5′-ACA GTT CCT TCA CCG TCT GG-3′ | 58 °C | |
| HK2 | 5′-CAA AGT GAC AGT GGG TGT GG-3′ | 5′-GCC AGG TCC TTC ACT GTC TC-3′ | 58 °C | |
| G6PDH | 5′-ACA TGA ATG CCC TCC ACC TG-3′ | 5′- GGT AGT GGT CGA TGC GGT AG-3′ | 56 °C | |
| RPLPO | 5′-CTG TCT GCA GAT TGG CTA CCC-3′ | 5′-GAT GGA TCA GCC AAG AAG GC-3′ | Genbank Accession No: NM_001002.3 | 56 °C |
| Western Blot (WB) | Dilution | Catalogue Number | Manufacturer |
|---|---|---|---|
| Primary Antibodies (WB) | |||
| AMPKα | 1:1000 | #2603 | Cell Signaling Technologies |
| p-AMPKα | 1:1000 | #2535 | Cell Signaling Technologies |
| GAPDH | 1:2000 | #sc-48167 | Santa-Cruz |
| MCT4 | 1:500 | #sc-50329 | Santa-Cruz |
| mTOR | 1:1000 | #2983 | Cell Signaling Technologies |
| p-mTOR | 1:1000 | #5536 | Cell Signaling Technologies |
| STAT3 | 1:1000 | #9145 | Cell Signaling Technologies |
| p-STAT3 (Y705) | 1:1000 | #9132 | Cell Signaling Technologies |
| p-STAT3 (S727) | 1:1000 | #9134 | Cell Signaling Technologies |
| Secondary Antibodies (WB) | |||
| mouse anti-rabbit IgG-HRP | 1:5000 | #sc-2357 | Santa-Cruz |
| goat anti-rabbit IgG-HRP | 1:5000 | #R-05072-500 | Advansta |
| donkey anti-goat IgG-HRP | 1:5000 | #sc-2020 | Santa-Cruz |
Appendix A.1. Supplementary Methods
Appendix A.1.1. Transient Transfection with siMCT4
Appendix A.1.2. High-Resolution Respirometry
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2018, 33, 152. [Google Scholar] [CrossRef] [Green Version]
- Fayzullin, A.; Sandberg, C.J.; Spreadbury, M.; Saberniak, B.M.; Grieg, Z.; Skaga, E.; Langmoen, I.A.; Vik-Mo, E.O. Phenotypic and expressional heterogeneity in the invasive glioma cells. Transl. Oncol. 2019, 12, 122–133. [Google Scholar] [CrossRef]
- Zhang, J.; Goliwas, K.F.; Wang, W.; Taufalele, P.V.; Bordeleau, F.; Reinhart-King, C.A. Energetic regulation of coordinated leader-follower dynamics during collective invasion of breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7867–7872. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; Vassallo, I.; Lanz, B.; Poitry-Yamate, C.; Hamou, M.F.; Cudalbu, C.; Gruetter, R.; Hegi, M.E. In vivo characterization of brain metabolism by (1) h MRS, (13) c MRS and (18) FDG PET reveals significant glucose oxidation of invasively growing glioma cells. Int. J. Cancer 2018, 143, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Gerthofer, V.; Kreutz, M.; Renner, K.; Jachnik, B.; Dettmer, K.; Oefner, P.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Combined modulation of tumor metabolism by metformin and diclofenac in glioma. Int. J. Mol. Sci. 2018, 19, 2586. [Google Scholar] [CrossRef] [Green Version]
- Seliger, C.; Meyer, A.L.; Renner, K.; Leidgens, V.; Moeckel, S.; Jachnik, B.; Dettmer, K.; Tischler, U.; Gerthofer, V.; Rauer, L.; et al. Metformin inhibits proliferation and migration of glioblastoma cells independently of TGF-beta2. Cell Cycle 2016, 15, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Bednarska, K.; Kus, P.; Fecka, I. Investigation of the phytochemical composition, antioxidant activity, and methylglyoxal trapping effect of galega officinalis l. Herb in vitro. Molecules 2020, 25, 5810. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Adeberg, S.; Bernhardt, D.; Ben Harrabi, S.; Bostel, T.; Mohr, A.; Koelsche, C.; Diehl, C.; Rieken, S.; Debus, J. Metformin influences progression in diabetic glioblastoma patients. Strahlenther. Onkol. 2015, 191, 928–935. [Google Scholar] [CrossRef]
- Seliger, C.; Genbrugge, E.; Gorlia, T.; Chinot, O.; Stupp, R.; Nabors, B.; Weller, M.; Hau, P.; EORTC Brain Tumor Group. Use of metformin and outcome of patients with newly diagnosed glioblastoma: Pooled analysis. Int. J. Cancer 2020, 146, 803–809. [Google Scholar] [CrossRef]
- Seliger, C.; Luber, C.; Gerken, M.; Schaertl, J.; Proescholdt, M.; Riemenschneider, M.J.; Meier, C.R.; Bogdahn, U.; Leitzmann, M.F.; Klinkhammer-Schalke, M.; et al. Use of metformin and survival of patients with high-grade glioma. Int. J. Cancer 2019, 144, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Welch, M.R.; Grommes, C. Retrospective analysis of the effects of steroid therapy and antidiabetic medication on survival in diabetic glioblastoma patients. CNS Oncol. 2013, 2, 237–246. [Google Scholar] [CrossRef]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef]
- Wurth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Leidgens, V.; Proske, J.; Rauer, L.; Moeckel, S.; Renner, K.; Bogdahn, U.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Stattic and metformin inhibit brain tumor initiating cells by reducing STAT3-phosphorylation. Oncotarget 2017, 8, 8250–8263. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhou, P.; Xu, K.; Chen, T.; Jiao, J.; Wei, H.; Yang, X.; Xu, W.; Wan, W.; Xiao, J. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 2020, 16, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Moeckel, S.; Vollmann-Zwerenz, A.; Proescholdt, M.; Brawanski, A.; Riemenschneider, M.J.; Bogdahn, U.; Bosserhoff, A.K.; Spang, R.; Hau, P. Validation study: Response-predictive gene expression profiling of glioma progenitor cells in vitro. PLoS ONE 2016, 11, e0151312. [Google Scholar] [CrossRef]
- Brown, D.V.; Daniel, P.M.; D’Abaco, G.M.; Gogos, A.; Ng, W.; Morokoff, A.P.; Mantamadiotis, T. Coexpression analysis of CD133 and CD44 identifies proneural and mesenchymal subtypes of glioblastoma multiforme. Oncotarget 2015, 6, 6267–6280. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. Emt: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [Green Version]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-mesenchymal transition: Phenotypic plasticity to acquire multitherapy resistance in glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, N.; Delbridge, C.; Gempt, J.; Feuchtinger, A.; Walch, A.; Schirmer, L.; Bunk, W.; Aschenbrenner, T.; Liesche-Starnecker, F.; Schlegel, J. The intratumoral heterogeneity reflects the intertumoral subtypes of glioblastoma multiforme: A regional immunohistochemistry analysis. Front. Oncol. 2020, 10, 494. [Google Scholar] [CrossRef]
- Guo, H.; Nan, Y.; Zhen, Y.; Zhang, Y.; Guo, L.; Yu, K.; Huang, Q.; Zhong, Y. miRNA-451 inhibits glioma cell proliferation and invasion by downregulating glucose transporter 1. Tumour Biol. 2016, 37, 13751–13761. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; et al. Lactate promotes glioma migration by TGF beta2-dependent regulation of matrix metalloproteinase-2. Neuro-Oncology 2009, 11, 368–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakovic, A.; Harhaji, L.; Stevanovic, D.; Markovic, Z.; Sumarac-Dumanovic, M.; Starcevic, V.; Micic, D.; Trajkovic, V. Dual antiglioma action of metformin: Cell cycle arrest and mitochondria-dependent apoptosis. Cell. Mol. Life Sci. 2007, 64, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Yi, Y.; Liu, Y.; Liu, X.; Keller, E.T.; Qian, C.N.; Zhang, J.; Lu, Y. Metformin targets multiple signaling pathways in cancer. Chin. J. Cancer 2017, 36, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Nakano, I. Therapeutic potential of targeting glucose metabolism in glioma stem cells. Expert Opin. Ther. Targets 2014, 18, 1233–1236. [Google Scholar] [CrossRef] [Green Version]
- Voss, D.M.; Spina, R.; Carter, D.L.; Lim, K.S.; Jeffery, C.J.; Bar, E.E. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci. Rep. 2017, 7, 4292. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.S.; Lim, K.J.; Price, A.C.; Orr, B.A.; Eberhart, C.G.; Bar, E.E. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene 2014, 33, 4433–4441. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018, 25, 3047–3058.e3044. [Google Scholar] [CrossRef] [Green Version]
- Hamann, A.; Benecke, H.; Greten, H.; Matthaei, S. Metformin increases glucose transporter protein and gene expression in human fibroblasts. Biochem. Biophys. Res. Commun. 1993, 196, 382–387. [Google Scholar] [CrossRef]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [Green Version]
- Oizel, K.; Chauvin, C.; Oliver, L.; Gratas, C.; Geraldo, F.; Jarry, U.; Scotet, E.; Rabe, M.; Alves-Guerra, M.C.; Teusan, R.; et al. Efficient mitochondrial glutamine targeting prevails over glioblastoma metabolic plasticity. Clin. Cancer Res. 2017, 23, 6292–6304. [Google Scholar] [CrossRef] [Green Version]
- Cuyas, E.; Fernandez-Arroyo, S.; Corominas-Faja, B.; Rodriguez-Gallego, E.; Bosch-Barrera, J.; Martin-Castillo, B.; De Llorens, R.; Joven, J.; Menendez, J.A. Oncometabolic mutation IDH1 R132H confers a metformin-hypersensitive phenotype. Oncotarget 2015, 6, 12279–12296. [Google Scholar] [CrossRef] [Green Version]
- Gogolla, N.; Galimberti, I.; DePaola, V.; Caroni, P. Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nat. Protoc. 2006, 1, 1165–1171. [Google Scholar] [CrossRef]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef] [Green Version]
- Schulze, M.; Sommer, A.; Plotz, S.; Farrell, M.; Winner, B.; Grosch, J.; Winkler, J.; Riemenschneider, M.J. Sporadic parkinson’s disease derived neuronal cells show disease-specific mRNA and small RNA signatures with abundant deregulation of piRNAs. Acta Neuropathol. Commun. 2018, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Heinrich, P.; Kohler, C.; Ellmann, L.; Kuerner, P.; Spang, R.; Oefner, P.J.; Dettmer, K. Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with isocorrector. Sci. Rep. 2018, 8, 17910. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seliger, C.; Meyer, A.-L.; Leidgens, V.; Rauer, L.; Moeckel, S.; Jachnik, B.; Proske, J.; Dettmer, K.; Rothhammer-Hampl, T.; Kaulen, L.D.; et al. Metabolic Heterogeneity of Brain Tumor Cells of Proneural and Mesenchymal Origin. Int. J. Mol. Sci. 2022, 23, 11629. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911629
Seliger C, Meyer A-L, Leidgens V, Rauer L, Moeckel S, Jachnik B, Proske J, Dettmer K, Rothhammer-Hampl T, Kaulen LD, et al. Metabolic Heterogeneity of Brain Tumor Cells of Proneural and Mesenchymal Origin. International Journal of Molecular Sciences. 2022; 23(19):11629. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911629
Chicago/Turabian StyleSeliger, Corinna, Anne-Louise Meyer, Verena Leidgens, Lisa Rauer, Sylvia Moeckel, Birgit Jachnik, Judith Proske, Katja Dettmer, Tanja Rothhammer-Hampl, Leon D. Kaulen, and et al. 2022. "Metabolic Heterogeneity of Brain Tumor Cells of Proneural and Mesenchymal Origin" International Journal of Molecular Sciences 23, no. 19: 11629. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911629