

Cytoskeleton Elements Contribute to Prion Peptide-Induced Endothelial Barrier Breakdown in a Blood–Brain Barrier In Vitro System
 , , and
, , and 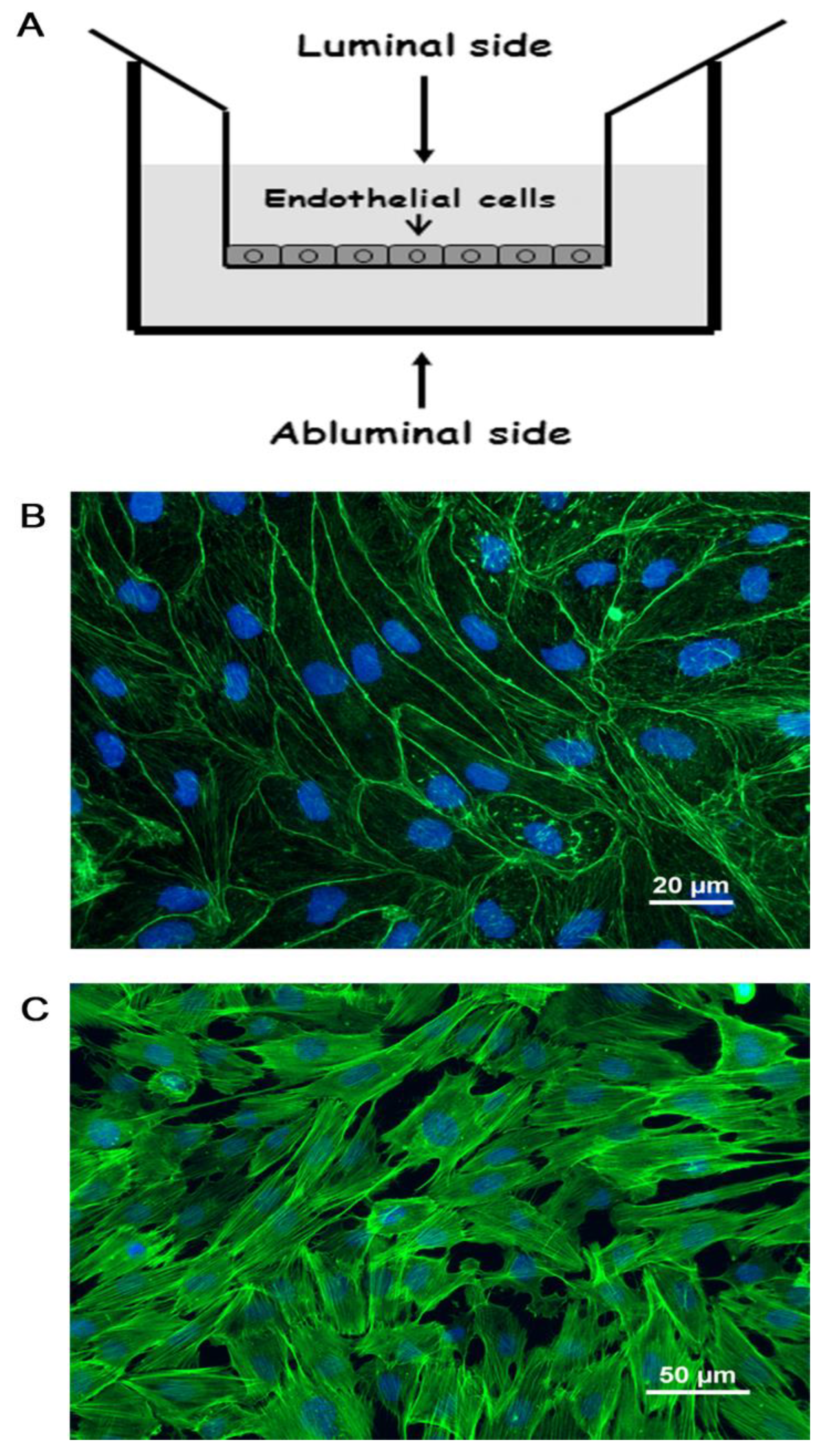{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Brain Capillary Endothelial Cells
2.2. PrP 106-126 Alters cAMP and Nitric Oxide Levels in Brain Endothelial Cells

2.3. PrP 106-126 Modulates the Activation State of the Small GTPases Rac1 and RhoA
2.4. Stress Fibers Formation and Cortactin Reduction in Brain Endothelial Cells Exposed to PrP 106-126
2.5. PrP 106-126 Increases Cytoskeleton-Related Proteins Expression in Brain Endothelial Cells
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Materials
4.3. Media
4.4. Peptides
4.5. Cells
4.6. In-Vitro BBB Model and Transendothelial Electrical Resistance Measurements
4.7. cAMP Determination
4.8. Nitrite Measurements
4.9. eNOS, iNOS and Cortactin Expression
4.10. Rac1 and RhoA Activation Assay
4.11. Immunocytochemistry
4.12. Liquid Chromatography-Mass Spectrometry (LC-MS)
4.12.1. Proteomic Analysis
4.12.2. Shotgun Proteomics
4.12.3. Targeted Proteomics—Selective Reaction Monitoring
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AJ | adherence junctions |
| BBB | blood-brain barrier |
| BCA | bicinchoninic acid |
| BEC | brain endothelial cells |
| GTPases | small guanosine triphosphatases |
| iNOS and eNOS | inducible and endothelial nitric oxide synthase |
| LC-MS | liquid chromatography-mass spectrometry |
| LPS | lipopolysaccharide |
| NO | nitric oxide |
| TJ | tight junction |
| PBS | phosphate-buffered saline |
| PrPC | cellular human prion protein |
| ROS | reactive oxygen species |
| TEER | trans-endothelial electrical resistance |
References
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [Green Version]
- Barisano, G.; Montagne, A.; Kisler, K.; Schneider, J.A.; Wardlaw, J.M.; Zlokovic, B.V. Blood brain barrier link to human cognitive impairment and Alzheimer’s disease. Nat. Cardiovasc. Res. 2022, 1, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [PubMed] [Green Version]
- Garcao, P.; Oliveira, C.R.; Agostinho, P. Comparative study of microglia activation induced by amyloid-beta and prion peptides: Role in neurodegeneration. J. Neurosci. Res. 2006, 84, 182–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [PubMed]
- Bicker, J.; Alves, G.; Fortuna, A.; Falcao, A. Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: A review. Eur. J. Pharm. Biopharm. 2014, 87, 409–432. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G. Endothelial tight junctions: Permeable barriers of the vessel wall. Thromb. Haemost. 2006, 95, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuhaus, J.; Risau, W.; Wolburg, H. Induction of blood-brain barrier characteristics in bovine brain endothelial cells by rat astroglial cells in transfilter coculture. Ann. N. Y. Acad. Sci. 1991, 633, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Niehoff, M.L.; Adessi, C.; Soto, C. Passage of murine scrapie prion protein across the mouse vascular blood-brain barrier. Biochem. Biophys. Res. Commun. 2004, 318, 125–130. [Google Scholar] [CrossRef]
- Banks, W.A.; Robinson, S.M.; Diaz-Espinoza, R.; Urayama, A.; Soto, C. Transport of prion protein across the blood-brain barrier. Exp. Neurol. 2009, 218, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandner, S.; Isenmann, S.; Kuhne, G.; Aguzzi, A. Identification of the end stage of scrapie using infected neural grafts. Brain Pathol. 1998, 8, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Cooper, I.; Cohen-Kashi Malina, K.; Cagnotto, A.; Bazzoni, G.; Salmona, M.; Teichberg, V.I. Interactions of the prion peptide (PrP 106–126) with brain capillary endothelial cells: Coordinated cell killing and remodeling of intercellular junctions. J. Neurochem. 2011, 116, 467–475. [Google Scholar] [CrossRef]
- Deli, M.A.; Sakaguchi, S.; Nakaoke, R.; Abraham, C.S.; Takahata, H.; Kopacek, J.; Shigematsu, K.; Katamine, S.; Niwa, M. PrP fragment 106–126 is toxic to cerebral endothelial cells expressing PrP(C). Neuroreport 2000, 11, 3931–3936. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, C.J.; Mahdavi, J.; Thornton, J.; Mann, B.; Wooldridge, K.G.; Abouseada, N.; Oldfield, N.J.; Self, T.; Ala’Aldeen, D.A.; Tuomanen, E.I. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Investig. 2009, 119, 1638–1646. [Google Scholar] [CrossRef] [Green Version]
- Poleshchuk, N.N.; I Votyakov, V.; Duboiskaya, G.P.; Grigoriev, D.G.; Kolomiets, N.D.; Yug, I. Structural and functional changes of blood-brain barrier and indication of prion amyloid filaments in experimental amyotrophic leucospongiosis. Acta. Virol. 1992, 36, 293–303. [Google Scholar]
- Song, K.; Han, H.J.; Kim, S.; Kwon, J. Thymosin beta 4 attenuates PrP(106–126)-induced human brain endothelial cells dysfunction. Eur. J. Pharmacol. 2020, 869, 172891. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Mark, A.E.; Saracino, G.A.; Cosentino, U.; Pitea, D.; Moro, G.; Salmona, M. Conformational polymorphism of the PrP106–126 peptide in different environments: A molecular dynamics study. J. Phys. Chem. B 2006, 110, 1423–1428. [Google Scholar] [CrossRef]
- Forloni, G.; Angeretti, N.; Chiesa, R.; Monzani, E.; Salmona, M.; Bugiani, O.; Tagliavini, F. Neurotoxicity of a prion protein fragment. Nature 1993, 362, 543–546. [Google Scholar] [CrossRef]
- Brandenburg, L.-O.; Koch, T.; Sievers, J.; Lucius, R. Internalization of PrP106–126 by the formyl-peptide-receptor-like-1 in glial cells. J. Neurochem. 2007, 101, 718–728. [Google Scholar] [CrossRef]
- McHattie, S.J.; Brown, D.R.; Bird, M.M. Cellular uptake of the prion protein fragment PrP106–126 in vitro. J. Neurocytol. 1999, 28, 149–159. [Google Scholar] [CrossRef]
- Tagliavini, F.; Prelli, F.; Porro, M.; Rossi, G.; Giaccone, G.; Farlow, M.R.; Dlouhy, S.R.; Ghetti, B.; Bugiani, O.; Frangione, B. Amyloid fibrils in Gerstmann-Straussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell 1994, 79, 695–703. [Google Scholar] [CrossRef]
- Tagliavini, F.; Lievens, P.M.; Tranchant, C.; Warter, J.M.; Mohr, M.; Giaccone, G.; Perini, F.; Rossi, G.; Salmona, M.; Piccardo, P.; et al. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Sträussler-Scheinker disease A117V. J. Biol. Chem. 2001, 276, 6009–6015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deli, M.A.; Abraham, C.S.; Kataoka, Y.; Niwa, M. Permeability studies on in vitro blood-brain barrier models: Physiology, pathology, and pharmacology. Cell. Mol. Neurobiol. 2005, 25, 59–127. [Google Scholar]
- Lorenowicz, M.J.; Fernandez-Borja, M.; Hordijk, P.L. cAMP signaling in leukocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, C.C.; Curry, F.E. Microvascular permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Nieuw Amerongen, G.P.; van Hinsbergh, V.W. Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vascul. Pharmacol. 2002, 39, 257–272. [Google Scholar] [PubMed]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kashi Malina, K.; Cooper, I.; Teichberg, V.I. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Res. 2009, 1284, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Franke, H.; Galla, H.; Beuckmann, C.T. Primary cultures of brain microvessel endothelial cells: A valid and flexible model to study drug transport through the blood-brain barrier in vitro. Brain Res. Brain Res. Protoc. 2000, 5, 248–256. [Google Scholar] [CrossRef]
- Poller, B.; Gutmann, H.; Krahenbuhl, S.; Weksler, B.; Romero, I.; Couraud, P.O.; Tuffin, G.; Drewe, J.; Huwyler, J. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J. Neurochem. 2008, 107, 1358–1368. [Google Scholar] [CrossRef]
- Stelzner, T.J.; Weil, J.V.; O’Brien, R.F. Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J. Cell. Physiol. 1989, 139, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Rist, R.J.; Romero, I.A.; Chan, M.W.; Couraud, P.O.; Roux, F.; Abbott, N.J. F-actin cytoskeleton and sucrose permeability of immortalised rat brain microvascular endothelial cell monolayers: Effects of cyclic AMP and astrocytic factors. Brain Res. 1997, 768, 10–18. [Google Scholar] [CrossRef]
- Rubin, L.L.; Hall, D.E.; Porter, S.; Barbu, K.; Cannon, C.; Horner, H.C.; Janatpour, M.; Liaw, C.W.; Manning, K.; Morales, J.; et al. A cell culture model of the blood-brain barrier. J. Cell. Biol. 1991, 115, 1725–1735. [Google Scholar] [CrossRef] [Green Version]
- Galea, E.; Feinstein, D.L. Regulation of the expression of the inflammatory nitric oxide synthase (NOS2) by cyclic AMP. FASEB J. 1999, 13, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veszelka, S.; Pasztoi, M.; Farkas, A.E.; Krizbai, I.; Ngo, T.K.; Niwa, M.; Abraham, C.S.; Deli, M.A. Pentosan polysulfate protects brain endothelial cells against bacterial lipopolysaccharide-induced damages. Neurochem. Int. 2007, 50, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, N.; Waschke, J. Impaired cAMP and Rac 1 signaling contribute to TNF-alpha-induced endothelial barrier breakdown in microvascular endothelium. Microcirculation 2009, 16, 521–533. [Google Scholar] [CrossRef]
- Schlegel, N.; Baumer, Y.; Drenckhahn, D.; Waschke, J. Lipopolysaccharide-induced endothelial barrier breakdown is cyclic adenosine monophosphate dependent in vivo and in vitro. Crit. Care Med. 2009, 37, 1735–1743. [Google Scholar] [CrossRef]
- Arce, F.T.; Whitlock, J.L.; Birukova, A.A.; Birukov, K.G.; Arnsdorf, M.F.; Lal, R.; Garcia, J.G.; Dudek, S.M. Regulation of the micromechanical properties of pulmonary endothelium by S1P and thrombin: Role of cortactin. Biophys. J. 2008, 95, 886–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Alekseeva, E.; Mikaelyan, A.; Birukov, K.G. HGF attenuates thrombin-induced endothelial permeability by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the Rho pathway. FASEB J. 2007, 21, 2776–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, N.; Burger, S.; Golenhofen, N.; Walter, U.; Drenckhahn, D.; Waschke, J. The role of VASP in regulation of cAMP- and Rac 1-mediated endothelial barrier stabilization. Am. J. Physiol. Cell Physiol. 2008, 294, C178–C188. [Google Scholar] [CrossRef] [PubMed]
- Waschke, J.; Burger, S.; Curry, F.R.; Drenckhahn, D.; Adamson, R.H. Activation of Rac-1 and Cdc42 stabilizes the microvascular endothelial barrier. Histochem. Cell Biol. 2006, 125, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- Schreibelt, G.; Kooij, G.; Reijerkerk, A.; van Doorn, R.; Gringhuis, S.I.; van der Pol, S.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Piontek, J.; et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007, 21, 3666–3676. [Google Scholar] [CrossRef] [Green Version]
- Ziche, M.; Morbidelli, L. Nitric oxide and angiogenesis. J. Neurooncol. 2000, 50, 139–148. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, B.H.; Park, S.J.; Jin, J.K.; Jeon, Y.C.; Wen, G.Y.; Shin, H.Y.; Carp, R.I.; Kim, Y.S. Association of endothelial nitric oxide synthase and mitochondrial dysfunction in the hippocampus of scrapie-infected mice. Hippocampus 2011, 21, 319–333. [Google Scholar] [CrossRef]
- Romero, I.A.; Radewicz, K.; Jubin, E.; Michel, C.C.; Greenwood, J.; Couraud, P.O.; Adamson, P. Changes in cytoskeletal and tight junctional proteins correlate with decreased permeability induced by dexamethasone in cultured rat brain endothelial cells. Neurosci. Lett. 2003, 344, 112–116. [Google Scholar] [CrossRef]
- Couty, J.P.; Rampon, C.; Leveque, M.; Laran-Chich, M.P.; Bourdoulous, S.; Greenwood, J.; Couraud, P.O. PECAM-1 engagement counteracts ICAM-1-induced signaling in brain vascular endothelial cells. J. Neurochem. 2007, 103, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Lambotin, M.; Hoffmann, I.; Laran-Chich, M.P.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Invasion of endothelial cells by Neisseria meningitidis requires cortactin recruitment by a phosphoinositide-3-kinase/Rac1 signalling pathway triggered by the lipo-oligosaccharide. J. Cell. Sci. 2005, 118, 3805–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, T.; Pascual, A. Identification of genes differentially expressed in SH-SY5Y neuroblastoma cells exposed to the prion peptide 106–126. Eur. J. Neurosci. 2007, 26, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.; Roman, S.; Ratia, M.; Camps, P.; Munoz-Torrero, D.; Colombo, L.; Manzoni, C.; Salmona, M.; Badia, A.; Clos, M.V. Acetylcholinesterase triggers the aggregation of PrP 106–126. Biochem. Biophys. Res. Commun. 2006, 346, 89–94. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, I.; Cohen-Kashi Malina, K.; Levin, Y.; Gabashvili, A.; Mohar, B.; Cagnotto, A.; Salmona, M.; Teichberg, V.I. Cytoskeleton Elements Contribute to Prion Peptide-Induced Endothelial Barrier Breakdown in a Blood–Brain Barrier In Vitro System. Int. J. Mol. Sci. 2022, 23, 12126. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012126
Cooper I, Cohen-Kashi Malina K, Levin Y, Gabashvili A, Mohar B, Cagnotto A, Salmona M, Teichberg VI. Cytoskeleton Elements Contribute to Prion Peptide-Induced Endothelial Barrier Breakdown in a Blood–Brain Barrier In Vitro System. International Journal of Molecular Sciences. 2022; 23(20):12126. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012126
Chicago/Turabian StyleCooper, Itzik, Katayun Cohen-Kashi Malina, Yishai Levin, Alexandra Gabashvili, Boaz Mohar, Alfredo Cagnotto, Mario Salmona, and Vivian I. Teichberg. 2022. "Cytoskeleton Elements Contribute to Prion Peptide-Induced Endothelial Barrier Breakdown in a Blood–Brain Barrier In Vitro System" International Journal of Molecular Sciences 23, no. 20: 12126. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012126