
The Contribution of JAK2 46/1 Haplotype in the Predisposition to Myeloproliferative Neoplasms

 , , and
, , and
Abstract
:1. Introduction
2. Janus Kinase Gene (JAK2)
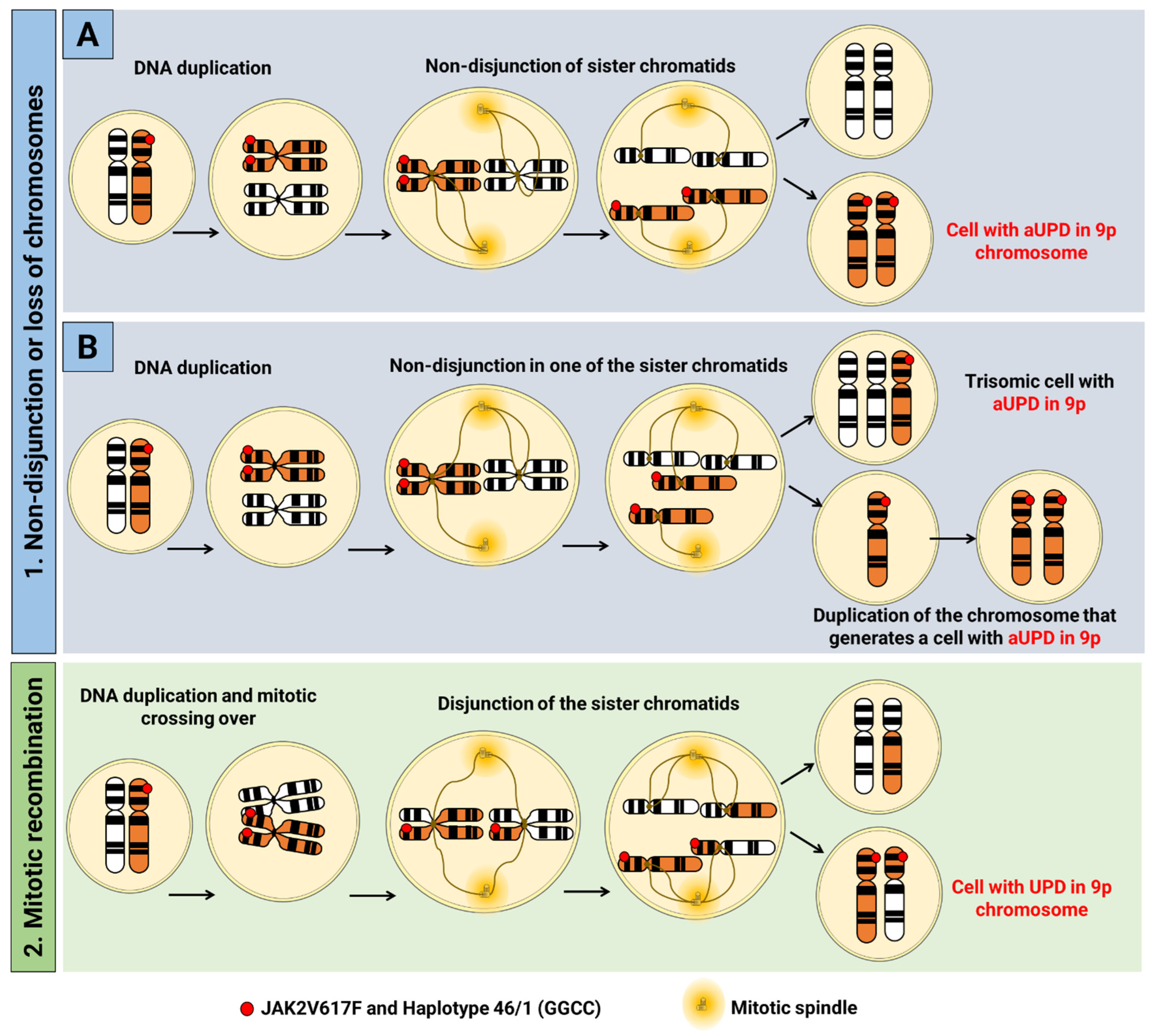
2.1. Acquired Uniparental Dysomy
2.2. JAK2V617F Variant
3. 46/1 Haplotype

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNV | References | Conclusions |
|---|---|---|
| rs10974944 | [68,76,89,97,98,99,100,101,102] | Studies carried out in populations of Brazilian, Japanese, and Chinese origin; this variant has a strong association with JAK2V617F positive MPN patients when compared to controls; rs10974944 (G) is a risk allele for MPNs. |
| rs12686652 | [89] | Significantly associated with patients with PV in this case-control study, but no association with MPNs in the Japanese population. |
| rs12335546 | ||
| rs12343867 | [71,89,90,99,100,101,102] | Associated with positive JAK2V617F in the populations of Japan, China, and Taiwan, especially in individuals with PV; this is used as a haplotype marker. Association with splenomegaly has been reported and is in LD with other SNVs of haplotype 46/1. |
| rs4495487 | [89] | More frequent in PV patients in a case-control study in Japan. It has not been reported in Caucasian populations and may contribute to the MPN phenotype in the Japanese population. |
| rs691857 | [101] | No significant association. |
| rs17803986 | ||
| rs7848509 | ||
| rs10758677 | ||
| rs3780367 | [103,104] | In linkage disequilibrium with other markers of the haplotype and has significant association with MPNs, but no population data. |
| rs12340895 | [100] | Associated with JAK2V617F positive MPNs in Chinese patients. |
| rs12342421 | [100] | Associated with the predisposition to develop JAK2V617F positive MPNs (OR = 3.55) in carriers for the minor C allele (in Chinese populations) with a 250% increased risk for disease, regardless of haplotype 46/1. |
| rs1159782 | [99,104] | It is in linkage disequilibrium with markers of the 46/1 haplotype. |
| rs10119004 | [100] | Associated with positive JAK2V617F and reported for the first time in the same study |
| rs12343065 | ||
| rs10815162 | ||
| rs7857730 | ||
| rs7847294 | ||
| rs3780378 | ||
| rs2149556 | ||
| rs2149555 | ||
| rs1887428 | [103] | Able to alter the expression rate of JAK2. |
4. Association between the 46/1 Haplotype and the JAK2V617F Variant
Haplotype 46/1 Agreement with JAK2V617F in Different Populations
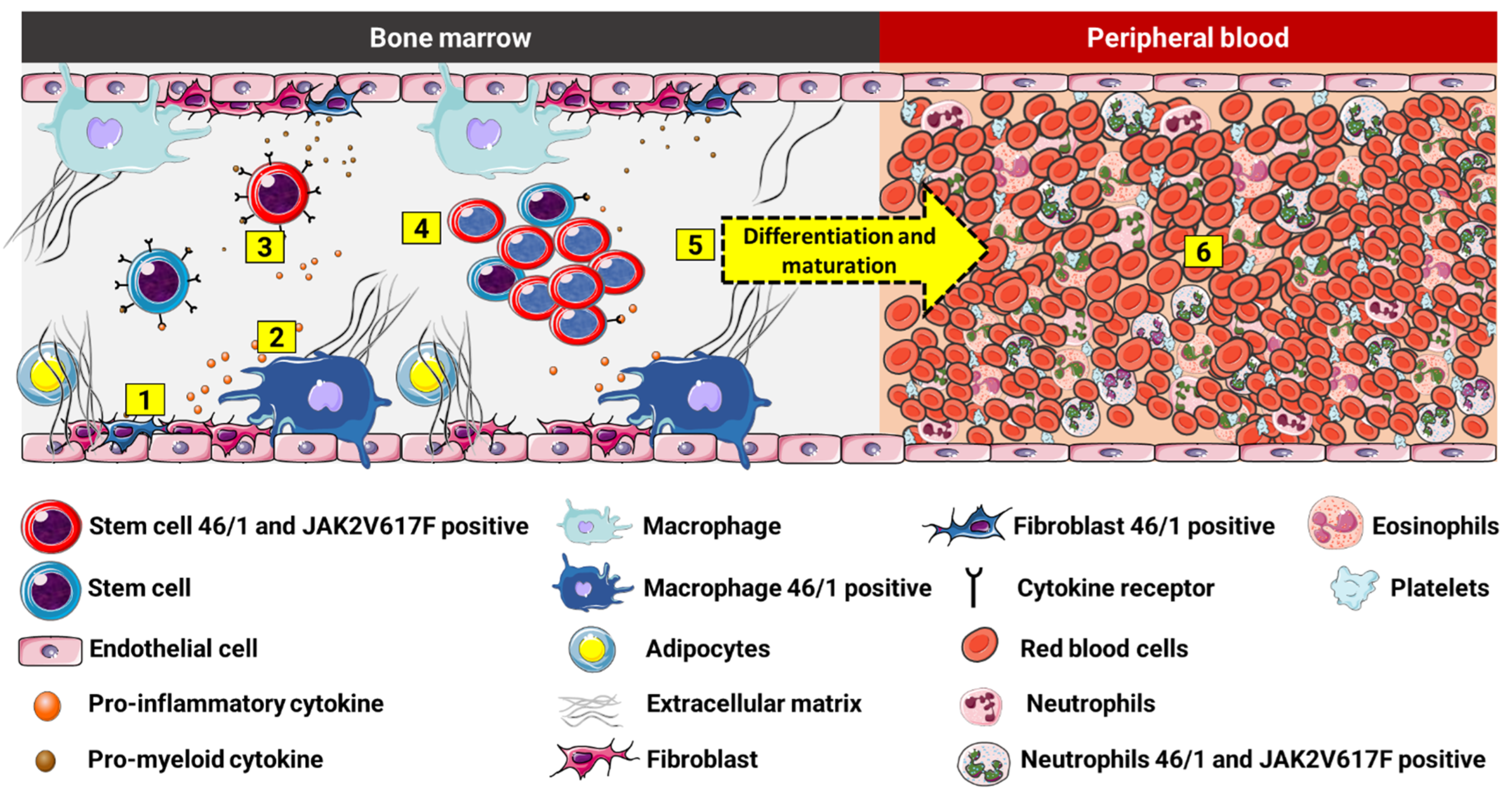
5. Contribution of 46/1 to Inflammatory Dysregulation in MPNs
6. Clinical and Laboratory Characteristics of MPNs Related to the 46/1 Haplotype
7. Inheritance of MPNs and the Relationship with the 46/1 Haplotype
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.; Kiiskinen, T.; Lareau, A.; et al. Inherited myeloproliferative neoplasm risk impacts hematopoietic stem cells. Nature 2021, 586, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Koschmieder, S.; Foltz, L.; Guglielmelli, P.; Flindt, T.; Koehler, M.; Mathias, J.; Komatsu, N.; Boothroyd, R.N.; Spierer, A.; et al. The impact of myeloproliferative neoplasms (MPNs) on patient quality of life and productivity: Results from the international MPN Landmark survey. Ann. Hematol. 2017, 96, 1653–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbini, M.C.N.; Soares, F.A.; Morais, J.C.; Vassallo, J.; Pereira Velloso, E.D.R.; De Lourdes, L.F.; Chaufaille, M.; Chiattone, C.S.; Aldred, V.L.; Siqueira, S.A.C.; et al. Classificação dos tumores hematopoéticos e linfoides de acordo com a OMS: Padronização da nomenclatura em língua portuguesa, 4a edição. J. Bras. Patol. E Med. Lab. 2011, 47, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Abello, V.; Quintero, G.; Espinosa, D.; Solano, M.H.; Casas, C.P.; Saavedra, D.; Quintero, M.; Lobatón, J.F.; Sossa, C.; Peña, Á.; et al. Descripción de las características clínicas de las neoplasias mieloproliferativas crónicas (NMPC) Description of the clinical characteristics of chronic myeloproliferative neoplasms (MPNs) First report of the colombian registry of MPNs. Acta Médica Colomb. 2017, 42, 35–41. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Female Reproductive; World Health Organization: Geneva, Switzerland, 2014; ISBN 9789283224358.
- Cree, I.A. The WHO classification of haematolymphoid tumours. Leukemia 2022, 36, 1701–1702. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Bao, E.L.; Cheng, A.N.; Sankaran, V.G. The genetics of human hematopoiesis and its disruption in disease. EMBO Mol. Med. 2019, 11, e10316. [Google Scholar] [CrossRef]
- Spivak, J. Myeloproliferative neoplasms. Diagn. Histopathol. 2017, 27, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Baade, P.D.; Ross, D.M.; Anderson, L.A.; Forsyth, C.; Fritschi, L. Changing incidence of myeloproliferative neoplasms in Australia, 2003–2014. Am. J. Hematol. 2019, 94, E107–E109. [Google Scholar] [CrossRef] [Green Version]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Podoltsev, N.A.; Zeidan, A.M. Epidemiology of the classical myeloproliferative neoplasms: The four corners of an expansive and complex map. Blood Rev. 2020, 42, 100706. [Google Scholar] [CrossRef]
- Roaldsnes, C.; Holst, R.; Frederiksen, H.; Ghanima, W. Myeloproliferative neoplasms: Trends in incidence, prevalence and survival in Norway. Eur. J. Haematol. 2017, 98, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Lee, J.O.; Bang, S.M. Incidence, survival and prevalence statistics of classical myeloproliferative neoplasm in Korea. J. Korean Med. Sci. 2016, 31, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.A.; Choi, C.W. Recent insights regarding the molecular basis of myeloproliferative neoplasms. Korean J. Intern. Med. 2020, 35, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, C.; Immanuel, T.; Ruskova, A.; Theakston, E.; Kalev-Zylinska, M.L. The epidemiology of myeloproliferative neoplasms in new zealand between 2010 and 2017: Insights from the new zealand cancer registry. Curr. Oncol. 2021, 28, 1544–1557. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Zeidan, A.M.; Wang, R.; Podoltsev, N.A. Epidemiology of the philadelphia chromosome-negative classical myeloproliferative neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 177–189. [Google Scholar] [CrossRef]
- Jimenez, S. Neoplasias mieloproliferativas Myeloproliferative neoplasms. Acta Médica Colomb. 2017, 42, 15–17. [Google Scholar]
- Lima, J.G.; Lopes, R.; Barbieri, T. Perfil dos pacientes com neoplasia mieloproliferativa cromossomo philadelfia negativo na unidade de alta complexidade oncológica do Hospital São José em Criciúma/SC no período de 2008 a 2015. Arq. Catarin. Med. 2018, 34, 128–139. [Google Scholar]
- Chauffaille, M. de L.L.F. Neoplasias mieloproliferativas: Revisão dos critérios diagnósticos e dos aspectos clínicos. Rev. Bras. Hematol. Hemoter. 2010, 32, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Szuber, N.; Vallapureddy, R.R.; Penna, D.; Lasho, T.L.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; Tefferi, A. Myeloproliferative neoplasms in the young: Mayo Clinic experience with 361 patients age 40 years or younger. Am. J. Hematol. 2018, 93, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Burja, B.; Mertelj, T.; Frank-Bertoncelj, M. Hi-JAKi-ng synovial fibroblasts in inflammatory arthritis with JAK inhibitors. Front. Med. 2020, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gerds, A.T. Beyond JAK-STAT: Novel therapeutic targets in Ph-negative MPN. Am. Soc. Hematol. Educ. Progr. Book 2019, 2016, 407–414. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Verstovsek, S. Molecular pathways: JAK/STAT pathway: Mutations, inhibitors, and resistance. Clin. Cancer Res. 2013, 19, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Gilliland, D.G. Myeloproliferative disorders. Blood 2008, 112, 2190–2198. [Google Scholar] [CrossRef]
- Levine, R.L.; Pardanani, A.; Tefferi, A.; Gilliland, D.G. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 2007, 7, 673–683. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Homo Sapiens Janus Kinase 2 (JAK2), RefSeqGene (LRG_612) on Chromosome 9. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/nuccore/NG_009904.1?from=5001&to=147939&report=genbank (accessed on 9 February 2022).
- Catarsi, P.; Rosti, V.; Morreale, G.; Poletto, V.; Villani, L.; Bertorelli, R.; Pedrazzini, M.; Zorzetto, M.; Barosi, G. JAK2 exon 14 skipping in patients with primary myelofibrosis: A minor splice variant modulated by the JAK2-V617F allele burden. PLoS ONE 2015, 10, e0116636. [Google Scholar] [CrossRef]
- Santos, L.C.d. Estudo Citogenético E Pesquisa de Mutações Nos Genes JAK2 E MPL Em Policitemia Vera, Mielofibrose Primária e Trombocitemia Essencial; Universidade Federal de São Paulo: São Paulo, Brazil, 2010; p. 70. [Google Scholar]
- Freitas, M.C.d.S. Avaliação Do Papel da Proteína Tirosina-Kinase Janus Kinase 2 (jak-2) Em Modelo Murino de Lesão Hepática Induzida Por Isquemia De Perfusão; Universidade Federal de São Paulo: São Paulo, Brazil, 2010; Volume 2. [Google Scholar]
- Zhao, L.; Ma, Y.; Seemann, J.; Huang, L.J.S. A regulating role of the JAK2 FERM domain in hyperactivation of JAK2(V617F). Biochem. J. 2010, 426, 91–98. [Google Scholar] [CrossRef]
- Score, J.; Cross, N.C.P. Acquired uniparental disomy in myeloproliferative neoplasms. Hematol. Oncol. Clin. N. Am. 2012, 26, 981–991. [Google Scholar] [CrossRef]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Blood 2017, 130, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Engel, E. A new genetic concept: Uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 1980, 6, 137–143. [Google Scholar] [CrossRef]
- Erola, P.; Torabi, K.; Miró, R.; Camps, J. The non-random landscape of somatically-acquired uniparental disomy in cancer. Oncotarget 2019, 10, 3982–3984. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Maciejewski, J.P. Pathogenesis and consequences of uniparental disomy in cancer. Clin. Cancer Res. 2011, 17, 3913–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralovics, R.; Buser, A.S.; Teo, S.S.; Coers, J.; Tichelli, A.; Van der Maas, A.P.C. Comparison of molecular markers in a cohort of patients with chronic myeloproliferative disorders. Blood 2003, 102, 1869–1871. [Google Scholar] [CrossRef]
- Kralovics, R.; Guan, Y.; Prchal, J.T. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp. Hematol. 2002, 30, 229–236. [Google Scholar] [CrossRef]
- Song, J.; Shao, H. SNP array in hematopoietic neoplasms: A review. Microarrays 2015, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wheeler, D.A.; Prchal, J.T. Acquired uniparental disomy of chromosome 9p in hematologic malignancies. Exp. Hematol. 2016, 44, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Zoi, K.; Cross, N.C.P. Genomics of myeloproliferative neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef]
- Wang, K.; Swierczek, S.; Hickman, K.; Prchal, J.T.; Hakonarson, H. Convergent mechanisms of somatic mutations in polycythemia vera. Physiol. Behav. 2011, 12, 25–32. [Google Scholar] [CrossRef]
- Braunstein, E.M.; Moliterno, A.R. Back to biology: New insights on inheritance in myeloproliferative disorders. Curr. Hematol. Malig. Rep. 2014, 9, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; Cross, N.C.P. Inherited predisposition to myeloproliferative neoplasms. Ther. Adv. Hematol. 2013, 4, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomo, L.; Acha, P. Genetic aspects of myelodysplastic/myeloproliferative neoplasms. Cancers 2021, 3, 2120. [Google Scholar] [CrossRef]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Viny, A.D.; Levine, R.L. Genetics of myeloproliferative neoplasms. Cancer J. 2014, 20, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Skov, V. Next generation sequencing in MPNs. Lessons from the past and prospects for use as predictors of prognosis and treatment responses. Cancers 2020, 12, 2194. [Google Scholar] [CrossRef]
- Staerk, J.; Constantinescu, S.N. The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. JAK-STAT 2012, 1, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Bellanné-Chantelot, C.; Chaumarel, I.; Labopin, M.; Bellanger, F.; Barbu, V.; De Toma, C.; Delhommeau, F.; Casadevall, N.; Vainchenker, W.; Thomas, G.; et al. Genetic and clinical implications of the Val617Phe JAK2 mutation in 72 families with myeloproliferative disorders. Blood 2006, 108, 346–352. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.G.; Paes, J.; da Costa, A.G.; Malheiro, A.; Silva, G.V.; de Souza Mourão, L.P.; Tarragô, A.M. JAK2 variant signaling: Genetic, hematologic and immune implication in chronic myeloproliferative neoplasms. Biomolecules 2022, 12, 291. [Google Scholar] [CrossRef]
- González García, C.; Funes Vera, C.; Blanquer Blanquer, M.; Moraleda Jiménez, J.M. Síndromes mieloproliferativos. Med. Programa Form. Médica Contin. Acreditado 2012, 11, 1289–1297. [Google Scholar] [CrossRef]
- Di Battista, V.; Bochicchio, M.T.; Giordano, G.; Napolitano, M.; Lucchesi, A. Review genetics and pathogenetic role of inflammasomes in philadelphia negative chronic myeloproliferative neoplasms: A narrative review. Int. J. Mol. Sci. 2021, 22, 561. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2014, 369, 2391–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.H.; Lee, K.O.; Jang, J.H.; Jung, C.W.; Kim, J.W.; Kim, S.H.; Kim, H.J. High frequency of JAK2 exon 12 mutations in Korean patients with polycythaemia vera: Novel mutations and clinical significance. J. Clin. Pathol. 2016, 69, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Pietra, D.; Li, S.; Brisci, A.; Passamonti, F.; Rumi, E.; Theocharides, A.; Ferrari, M.; Gisslinger, H.; Kralovics, R.; Cremonesi, L.; et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 2008, 111, 1686–1689. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Avilés, L.; Besses, C.; Álvarez-Larrán, A.; Cervantes, F.; Hernández-Boluda, J.C.; Bellosillo, B. JAK2 exon 12 mutations in polycythemia vera or idiopathic erythrocytosis. Haematologica 2007, 92, 1717–1718. [Google Scholar] [CrossRef] [Green Version]
- Todorova, R.; Passweg, J.; Lundberg, P.; Tzankov, A. Does the order of mutational acquisition in myeloproliferative neoplasms matter? Evidence from JAK2 exon 12 and DNMT3A co-mutant polycythemia vera. J. Hematop. 2020, 13, 105–107. [Google Scholar] [CrossRef]
- Easwar, A.; Siddon, A.J. Genetic landscape of myeloproliferative neoplasms with an emphasis on molecular diagnostic laboratory testing. Life 2021, 11, 1158. [Google Scholar] [CrossRef] [PubMed]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Swierczek, S.I.; Hickman, K.; Walker, K.; Wang, K.; Drummond, J.; Doddapaneni, H.; Reid, J.G.; Muzny, D.M.; Gibbs, R.A.; et al. The relationship of JAK2V617F and acquired UPD at chromosome 9p. Leukemia 2014, 28, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini-e-Silva, S.; Santos, B.C.; Pereira, E.M.d.F.; Ferreira, M.E.; Baraldi, E.C.; Sell, A.M.; Visentainer, J.E.L. Evaluation of the association between the JAK2 46/1 haplotype and chronic myeloproliferative neoplasms in a Brazilian population. Clinics 2013, 68, 5–9. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Patnaik, M.M.; Finke, C.M.; Hussein, K.; Hogan, W.J.; Elliott, M.A.; Litzow, M.R.; Hanson, C.A.; Pardanani, A. JAK2 germline genetic variation affects disease susceptibility in primary myelofibrosis regardless of V617F mutational status: Nullizygosity for the JAK2 46/1 haplotype is associated with inferior survival. Leukemia 2010, 24, 105–109. [Google Scholar] [CrossRef]
- Landgren, O.; Goldin, L.R.; Kristinsson, S.Y.; Helgadottir, E.A.; Samuelsson, J.; Björkholm, M. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24 577 first-degree relatives of 11 039 patients with myeloproliferative neoplasms in Sweden. Blood 2008, 112, 2199–2204. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Guglielmelli, P. The JAK2 46/1 (GGCC) MPN-predisposing haplotype: A risky haplotype, after all. Am. J. Hematol. 2019, 94, 283–285. [Google Scholar] [CrossRef] [Green Version]
- Gou, P.; Zhang, W.; Giraudier, S. Insights into the potential mechanisms of JAK2V617F somatic mutation contributing distinct phenotypes in myeloproliferative neoplasms. Int. J. Mol. Sci. 2022, 23, 1013. [Google Scholar] [CrossRef]
- Yang, W.Y.; Hormozdiari, F.; Wang, Z.; He, D.; Pasaniuc, B.; Eskin, E. Leveraging reads that span multiple single nucleotide polymorphisms for haplotype inference from sequencing data. Bioinformatics 2013, 29, 2245–2252. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M. How many faces in MPDs ? Bad news for GVHD prevention. Blood 2008, 111, 2785. [Google Scholar] [CrossRef]
- Goldstein, D.B.; Cavalleri, G.L. NEWS & VIEWS Understanding human diversity. Nature 2005, 437, 5–6. [Google Scholar]
- Macedo, L.C.; Santos, B.C.; Pagliarini-e-Silva, S.; Pagnano, K.B.B.; Rodrigues, C.; Quintero, F.C.; Ferreira, M.E.; Baraldi, E.C.; Ambrosio-Albuquerque, E.P.; Sell, A.M.; et al. JAK2 46/1 haplotype is associated with JAK2 V617F-positive myeloproliferative neoplasms in Brazilian patients. Int. J. Lab. Hematol. 2015, 37, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Belmont, J.W.; Boudreau, A.; Leal, S.M.; Hardenbol, P.; Pasternak, S.; Wheeler, D.A.; Willis, T.D.; Yu, F.; Yang, H.; Gao, Y.; et al. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar] [CrossRef] [Green Version]
- Slatkin, M. Linkage disequilibrium—Understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, B. Linkage disequilibrium. Encycl. Bioinform. Comput. Biol. ABC Bioinform. 2018, 1–3, 763–765. [Google Scholar] [CrossRef]
- Neale, B.M. Introduction to linkage disequilibrium, the HapMap, and imputation. Cold Spring Harb. Protoc. 2010, 5, 10–13. [Google Scholar] [CrossRef]
- Tanaka, T. International HapMap project. Nippon Rinsho. Jpn. J. Clin. Med. 2005, 63 (Suppl. 1), 29–34. [Google Scholar] [CrossRef]
- International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 2005, 449, 851–861. [Google Scholar]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56. [Google Scholar] [CrossRef]
- Glusman, G.; Cox, H.C.; Roach, J.C. Whole-genome haplotyping approaches and genomic medicine. Genome Med. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Trillos, A.; Maffioli, M.; Colomer, D.; Alvarez-Larrán, A.; Pereira, A.; Angona, A.; Bellosillo, B.; Cervantes, F. Relationship between the 46/1 haplotype of the JAK2 gene and the JAK2 mutational status and allele burden, the initial findings, and the survival of patients with myelofibrosis. Ann. Hematol. 2014, 93, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Fridley, B.L.; Lasho, T.L.; Gilliland, D.G.; Tefferi, A. Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood 2008, 111, 2785–2789. [Google Scholar] [CrossRef] [PubMed]
- Masselli, E.; Pozzi, G.; Carubbi, C.; Vitale, M. The genetic makeup of myeloproliferative neoplasms: Role of germline variants in defining disease risk, phenotypic diversity and outcome. Cells 2021, 10, 2597. [Google Scholar] [CrossRef] [PubMed]
- Ohyashiki, J.H.; Yoneta, M.; Hisatomi, H.; Iwabuchi, T.; Umezu, T.; Ohyashiki, K. The C allele of JAK2 rs4495487 is an additional candidate locus that contributes to myeloproliferative neoplasm predisposition in the Japanese population. BMC Med. Genet. 2012, 13, 6. [Google Scholar] [CrossRef]
- Chiang, Y.; Chang, Y.; Lin, H.; Huang, L. Germline variations at JAK2, TERT, HBS1L-MYB and MECOM and the risk of myeloproliferative neoplasms in Taiwanese population. Oncotarget 2017, 8, 76204–76213. [Google Scholar] [CrossRef]
- Smalberg, J.H.; Koehler, E.; Murad, S.D.; Plessier, A.; Seijo, S.; Trebicka, J.; Primignani, M.; De Maat, M.P.M.; Garcia-Pagan, J.C.; Valla, D.C.; et al. The JAK2 46/1 haplotype in Budd-Chiari syndrome and portal vein thrombosis. Blood 2011, 117, 3968–3973. [Google Scholar] [CrossRef]
- Bak, M.; Jess, T.; Flachs, E.M.; Zwisler, A.D.; Juel, K.; Frederiksen, H. Risk of inflammatory bowel disease in patients with chronic myeloproliferative neoplasms: A danish nationwide cohort study. Cancers 2020, 12, 2700. [Google Scholar] [CrossRef]
- Zhang, J.X.; Song, J.; Wang, J.; Dong, W.G. JAK2 rs10758669 polymorphisms and susceptibility to ulcerative colitis and Crohn’s disease: A meta-analysis. Inflammation 2014, 37, 793–800. [Google Scholar] [CrossRef]
- Ferguson, L.R.; Han, D.Y.; Fraser, A.G.; Huebner, C.; Lam, W.J.; Morgan, A.R.; Duan, H.; Karunasinghe, N. Genetic factors in chronic inflammation: Single nucleotide polymorphisms in the STAT-JAK pathway, susceptibility to DNA damage and Crohn’s disease in a New Zealand population. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2010, 690, 108–115. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrikovics, H.; Nahajevszky, S.; Koszarska, M.; Meggyesi, N.; Bors, A.; Halm, G.; Lueff, S.; Lovas, N.; Matrai, Z.; Csomor, J.; et al. JAK2 46/1 haplotype analysis in myeloproliferative neoplasms and acute myeloid leukemia. Leukemia 2010, 24, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Hermouet, S.; Vilaine, M. The JAK2 46/1 haplotype: A marker of inappropriate myelomonocytic response to cytokine stimulation, leading to increased risk of inflammation, myeloid neoplasm, and impaired defense against infection? Haematologica 2011, 96, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Kilpivaara, O.; Mukherjee, S.; Schram, A.M.; Wadleigh, M.; Mullally, A.; Ebert, B.L.; Bass, A.; Marubayashi, S.; Heguy, A.; Garcia-Manero, G.; et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2V617F -positive myeloproliferative neoplasms. Nat. Genet. 2009, 41, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Li, S.L.; Zhang, P.J.; Sun, G.X.; Lu, Z.J. The JAK2 46/1 haplotype (GGCC) in myeloproliferative neoplasms and splanchnic vein thrombosis: A pooled analysis of 26 observational studies. Ann. Hematol. 2014, 93, 1845–1852. [Google Scholar] [CrossRef]
- Koh, S.P.; Yip, S.P.; Lee, K.K.; Chan, C.C.; Lau, S.M.; Kho, C.S.; Lau, C.K.; Lin, S.Y.; Lau, Y.M.; Wong, L.G.; et al. Genetic association between germline JAK2 polymorphisms and myeloproliferative neoplasms in Hong Kong Chinese population: A case-control study. BMC Genet. 2014, 15, 147. [Google Scholar] [CrossRef] [Green Version]
- Olcaydu, D.; Harutyunyan, A.; Jäger, R.; Berg, T.; Gisslinger, B.; Pabinger, I.; Gisslinger, H.; Kralovics, R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat. Genet. 2009, 41, 450–454. [Google Scholar] [CrossRef]
- Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. The JAK2 GGCC (46/1) haplotype in myeloproliferative neoplasms: Causal or random? Int. J. Mol. Sci. 2018, 19, 1152. [Google Scholar] [CrossRef] [Green Version]
- Schram, A.M.; Xu, X.; Kilpivaara, O.; Mukherjee, S.; Viny, A.D.; Guryanova, O.; Klein, R.J.; Levine, R.L. Genetic and functional investigation of germline JAK2 alleles that predispose to myeloproliferative neoplasms. Blood 2011, 118, 124. [Google Scholar] [CrossRef]
- IGSR|Populations. Available online: https://www.internationalgenome.org/data-portal/population (accessed on 9 February 2022).
- Rs10974944 (SNP)-Population Genetics-Homo_Sapiens-Ensembl Genome Browser 105. Available online: http://www.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=9:5070331-5071331;v=rs10974944;vdb=variation;vf=731581676 (accessed on 9 February 2022).
- Rs3780367 (SNP)-Genética de Populações-Homo_Sapiens-Ensembl Genoma Browser 105. Available online: http://www.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=9:5068255-5069255;v=rs3780367;vdb=variation;vf=729888189 (accessed on 9 February 2022).
- Rs17302090 (SNP)-Population Genetics-Homo_Sapiens-Ensembl Genome Browser 105. Available online: http://www.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=9:5073689-5074689;v=rs12343867;vdb=variation;vf=732179982 (accessed on 9 February 2022).
- Rs1159782 (SNP)-Population Genetics-Homo_Sapiens-Ensembl Genome Browser 105. Available online: http://www.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=9:5077617-5078617;v=rs1159782;vdb=variation;vf=729146257#population_freq_SAS (accessed on 9 February 2022).
- Tashi, T.; Swierczek, S.; Prchal, J.T. Familial MPN predisposition. Curr. Hematol. Malig. Rep. 2017, 12, 442–447. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. The germline JAK2 GGCC (46/1) haplotype and survival among 414 molecularly-annotated patients with primary myelofibrosis. Am. J. Hematol. 2019, 94, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Lanikova, L.; Babosova, O.; Prchal, J.T. Experimental modeling of myeloproliferative neoplasms. Genes 2019, 10, 813. [Google Scholar] [CrossRef] [Green Version]
- Trifa, A.P.; Bănescu, C.; Bojan, A.S.; Voina, C.M.; Popa, Ș.; Vișan, S.; Ciubean, A.D.; Tripon, F.; Dima, D.; Popov, V.M.; et al. MECOM, HBS1L-MYB, THRB-RARB, JAK2, and TERT polymorphisms defining the genetic predisposition to myeloproliferative neoplasms: A study on 939 patients. Am. J. Hematol. 2017, 93, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hu, T.; Wu, Z.; Kang, Z.; Liu, W.; Guan, M. The JAK2 46/1 haplotype is a risk factor for myeloproliferative neoplasms in Chinese patients. Int. J. Hematol. 2012, 96, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J. Somatic and germline genetics at the JAK2 locus. Nat. Methods 2009, 41, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Olcaydu, D.; Rumi, E.; Harutyunyan, A.; Passamonti, F.; Pietra, D.; Pascutto, C.; Berg, T.; Jäger, R.; Hammond, E.; Cazzola, M.; et al. The role of the JAK2 GGCC haplotype and the TET2 gene in familial myeloproliferative neoplasms. Haematologica 2011, 96, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.H.; Qu, C.K.; Pauly, M. Germline mutations in the bone marrow microenvironment and dysregulated hematopoiesis. Exp. Hematol. 2018, 66, 17–26. [Google Scholar] [CrossRef]
- Nahajevszky, S.; Andrikovics, H.; Batai, A.; Adam, E.; Bors, A.; Csomor, J.; Gopcsa, L.; Koszarska, M.; Kozma, A.; Lovas, N.; et al. The prognostic impact of germline 46/1 haplotype of Janus Kinase 2 in cytogenetically normal acute myeloid leukemia. Haematologica 2011, 96, 1613–1618. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Shao, M.; Peng, H.; Bi, Z.; Su, Z.; Li, H. In vitro differentiation of bone marrow stromal cells into neurons and glial cells and differential protein expression in a two-compartment bone marrow stromal cell/neuron co-culture system. J. Clin. Neurosci. 2010, 17, 908–913. [Google Scholar] [CrossRef]
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef]
- Nasillo, V.; Riva, G.; Paolini, A.; Forghieri, F.; Roncati, L.; Lusenti, B.; Maccaferri, M.; Messerotti, A.; Pioli, V.; Gilioli, A.; et al. Inflammatory microenvironment and specific t cells in myeloproliferative neoplasms: Immunopathogenesis and novel immunotherapies. Int. J. Mol. Sci. 2021, 22, 1906. [Google Scholar] [CrossRef] [PubMed]
- Manshouri, T.; Estrov, Z.; Quintás-Cardama, A.; Burger, J.; Zhang, Y.; Livun, A.; Knez, L.; Harris, D.; Creighton, C.J.; Kantarjian, H.M.; et al. Bone marrow stroma-secreted cytokines protect JAK2V617F-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. 2011, 71, 3831–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The molecular drivers of disease initiation, progression and transformation and their effect on treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Z.; Liu, L.; Gale, R.P.; Cross, N.C.P.; Jones, A.V.; Qin, T.; Ai, X.; Xu, J.; Zhang, T.; et al. JAK2V617F allele burden, JAK2 46/1 haplotype and clinical features of Chinese with myeloproliferative neoplasms. Leukemia 2013, 27, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, G.; Zhang, P.; Zhang, J.; Gui, E.; Zu, M.; Jia, E.; Xu, H.; Xu, L.; Zhang, J.; et al. JAK2 V617F mutation and 46/1 haplotype in Chinese Budd-Chiari syndrome patients. J. Gastroenterol. Hepatol. 2014, 29, 208–214. [Google Scholar] [CrossRef]
- Oddsson, A.; Kristinsson, S.Y.; Helgason, H.; Gudbjartsson, D.F.; Masson, G.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Steingrimsdottir, H.; Vidarsson, B.; et al. The germline sequence variant rs2736100-C in TERT associates with myeloproliferative neoplasms. Leukemia 2014, 28, 1371–1374. [Google Scholar] [CrossRef] [Green Version]
- Motazedi, E.; Maliepaard, C.; Finkers, R.; Visser, R.; De Ridder, D. Family-based haplotype estimation and allele dosage correction for polyploids using short sequence reads. Front. Genet. 2019, 10, 335. [Google Scholar] [CrossRef] [Green Version]
- Accurso, V.; Santoro, M.; Mancuso, S.; Vajana, G.; Tomasello, R.; Rotolo, C.; Camarda, G.; Mattana, M.; Siragusa, S. Familial essential thrombocythemia: 6 cases from a mono-institutional series. Clin. Case Rep. 2022, 10, e05525. [Google Scholar] [CrossRef]
- Skoda, R.C. Hereditary myeloproliferative disorders. Haematologica 2010, 95, 6–8. [Google Scholar] [CrossRef] [Green Version]
- Abruzzese, E.; Poeta, G.D.; Barbato, R.; Fratoni, S.; Trawinska, M.M.; Zangrilli, D.; Coletta, A.M.; Patroi, I.M.; Francesconi, F.; Santeusanio, G.; et al. Discordant distribution of JAK2 V617F mutation in siblings with familial. Blood 2015, 107, 4572–4574. [Google Scholar]
- Milosevic, J.D.; Kralovics, R. Genetic and epigenetic alterations of myeloproliferative disorders. Int. J. Hematol. 2013, 97, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, R.; Harutyunyan, A.S.; Rumi, E.; Pietra, D.; Berg, T.; Olcaydu, D.; Houlston, R.S.; Cazzola, M.; Kralovics, R. Common germline variation at the TERT locus contributes to familial clustering of myeloproliferative neoplasms. Am. J. Hematol. 2014, 89, 1107–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.V.; Campbell, P.J.; Beer, P.A.; Schnittger, S.; Vannucchi, A.M.; Zoi, K.; Percy, M.J.; McMullin, M.F.; Scott, L.M.; Tapper, W.; et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood 2010, 115, 4517–4523. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Gangat, N.; Caramazza, D.; Siragusa, S.; Hanson, C.A.; Pardanani, A.; Tefferi, A. MPL mutation effect on JAK2 46/1 haplotype frequency in JAK2V617F-negative myeloproliferative neoplasms. Leukemia 2010, 24, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Pietra, D.; Casetti, I.; Da Vià, M.C.; Elena, C.; Milanesi, C.; Rumi, E. JAK2 GGCC haplotype in MPL mutated myeloproliferative neoplasms. Am. J. Hematol. 2012, 87, 746–747. [Google Scholar] [CrossRef]
- Soler, G.; Bernal-Vicente, A.; Antón, A.I.; Torregrosa, J.M.; Caparrós-Pérez, E.; Sánchez-Serrano, I.; Martínez-Pérez, A.; Sánchez-Vega, B.; Vicente, V.; Ferrer-Marin, F. The JAK2 46/1 haplotype does not predispose to CALR-mutated myeloproliferative neoplasms. Ann. Hematol. 2015, 94, 789–794. [Google Scholar] [CrossRef]
- Gau, J.-P.; Chen, C.-C.; Liu, C.-J.; Yu, Y.-B.; Hsiao, L.-T.; Liu, J.-H.; Chen, P.-M.; Tzeng, C.-H. The 46/1 haplotype frequency is not increased in patients of essential thrombocythemia with CALR mutations. Blood 2014, 124, 5204. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in aging, inflammation, hematopoiesis and malignant transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- Balassa, K.; Krahling, T.; Remenyi, P.; Batai, A.; Bors, A.; Kiss, K.P.; Torbagyi, E.; Gopcsa, L.; Lengyel, L.; Barta, A.; et al. Recipient and donor JAK2 46/1 haplotypes are associated with acute graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Leuk. Lymphoma 2017, 58, 391–398. [Google Scholar] [CrossRef]
- Crawford, D.C.; Nickerson, D.A. Definition and clinical importance of haplotypes. Annu. Rev. Med. 2005, 56, 303–320. [Google Scholar] [CrossRef] [Green Version]
- Sud, A.; Chattopadhyay, S.; Thomsen, H.; Sundquist, K.; Sundquist, J.; Houlston, R.S.; Hemminki, K. Familial risks of acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative neoplasms. Blood 2018, 132, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Liu, D.; Cao, Z.; Zhu, S.; Li, H.; Su, H.; Zhang, L.; Xue, F.; Liu, X.; Zhang, X.; et al. Distinct molecular abnormalities underlie unique clinical features of essential thrombocythemia in children. Leukemia 2016, 30, 746–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Allison, D.B.; Hoeschele, I. Haplotyping methods for pedigrees. Hum. Hered. 2009, 67, 248–266. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.; Tian, Q. Systems approaches to biology and disease enable translational systems medicine. Genom. Proteom. Bioinform. 2012, 10, 181–185. [Google Scholar] [CrossRef]
- Hasan, S.; Cassinat, B.; Droin, N.; Le Couedic, J.P.; Favale, F.; Monte-Mor, B.; Lacout, C.; Fontenay, M.; Dosquet, C.; Chomienne, C.; et al. Use of the 46/1 haplotype to model JAK2 V617F clonal architecture in PV patients: Clonal evolution and impact of IFN treatment. Leukemia 2014, 28, 460–463. [Google Scholar] [CrossRef]
- Tan, J.; Chow, Y.P.; Zainul Abidin, N.; Chang, K.M.; Selvaratnam, V.; Tumian, N.R.; Poh, Y.M.; Veerakumarasivam, A.; Laffan, M.A.; Wong, C.L. Analysis of genetic variants in myeloproliferative neoplasms using a 22-gene next-generation sequencing panel. BMC Med. Genom. 2022, 15, 10. [Google Scholar] [CrossRef]
- Lee, J.E.; Choi, J.H.; Lee, J.H.; Lee, M.G. Gene SNPs and mutations in clinical genetic testing: Haplotype-based testing and analysis. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2005, 573, 195–204. [Google Scholar] [CrossRef]
- Kushekhar, K.; Paczesny, S. JAK polymorphisms: Jack of all cytokines, masters GVHD? Leuk. Lymphoma 2017, 58, 255–256. [Google Scholar] [CrossRef]





| MPN | Clinical Description | Epidemiology |
|---|---|---|
| Polycythemia vera (PV) | Unregulated proliferation of erythroid series elements and increased granulocyte and thrombocyte counts (panmyelosis) [4,5,7] | Incidence of 0.5–4.0 cases per 100,000 Australian individuals [10], Europeans [11,12], Koreans [13,14], New Zealanders [15], and North Americans [11] aged between 60 and 70 years [5,14,15,16,17,18] |
| Essential thrombocythemia (ET) | Elevated number of platelets in peripheral blood (>450 × 109/L), caused by megakaryocytic hyperplasia in the bone marrow, with alteration of other medullary sectors (erythrocytic or granulocytic) in a qualitative or quantitative way [4,5,19] | Affects individuals between the fifth and sixth decade of life with an incidence between 0.9–2.4 cases per 100,000 in North Americans [20], Koreans [13,14], and New Zealanders [14,15,20]. |
| Primary myelofibrosis (PMF) | MPN with a worse prognosis, characterized by the proliferation of predominantly abnormal megakaryocytes and granulocytes in the bone marrow, deposition of reticulin fibers, and extramedullary hematopoiesis [4,5,21] | Affects individuals between the sixth and seventh decade of life [5] and has an incidence of 0.33 cases per 100,000 individuals per year in North America [15]; 0.4 cases per 100,000 in the Republic of Korea [14]; and 0.88 cases per 100,000 individuals in New Zealand [15]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paes, J.; Silva, G.A.V.; Tarragô, A.M.; Mourão, L.P.d.S. The Contribution of JAK2 46/1 Haplotype in the Predisposition to Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2022, 23, 12582. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012582
Paes J, Silva GAV, Tarragô AM, Mourão LPdS. The Contribution of JAK2 46/1 Haplotype in the Predisposition to Myeloproliferative Neoplasms. International Journal of Molecular Sciences. 2022; 23(20):12582. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012582
Chicago/Turabian StylePaes, Jhemerson, George A. V. Silva, Andréa M. Tarragô, and Lucivana P. de Souza Mourão. 2022. "The Contribution of JAK2 46/1 Haplotype in the Predisposition to Myeloproliferative Neoplasms" International Journal of Molecular Sciences 23, no. 20: 12582. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012582