
Computational Modeling and Imaging of the Intracellular Oxygen Gradient
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
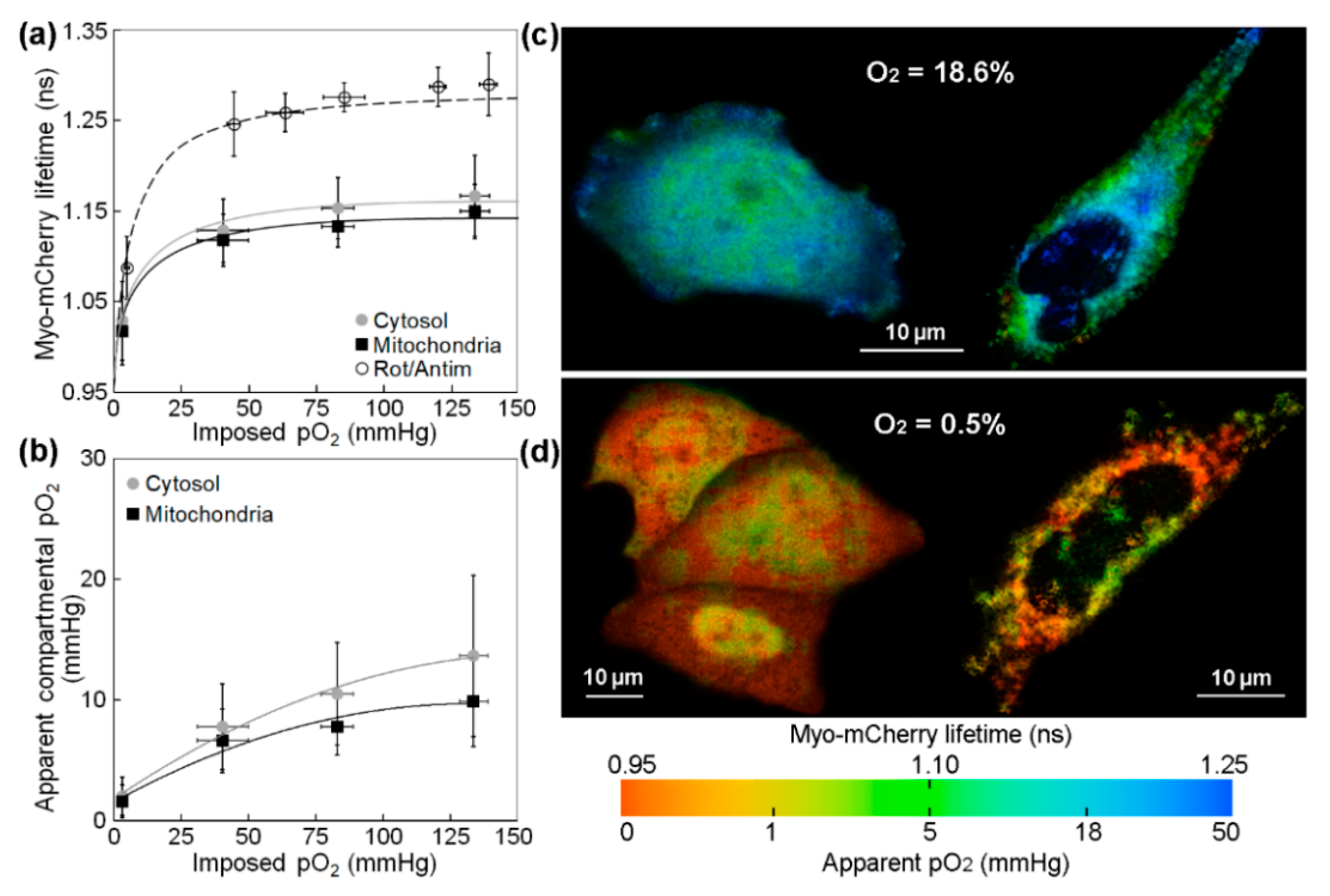
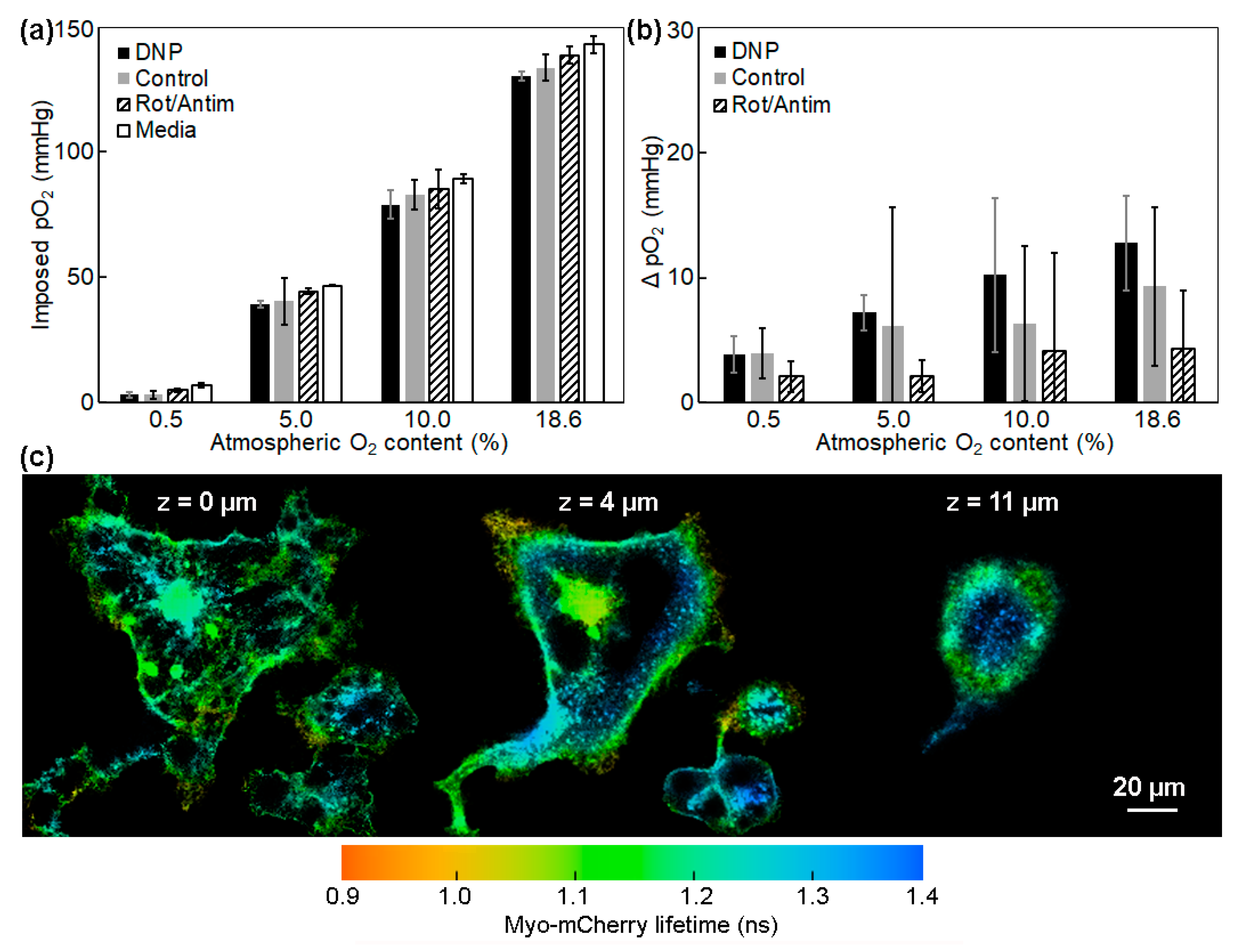
2.1. Measurements of the Intracellular pO2 Gradients
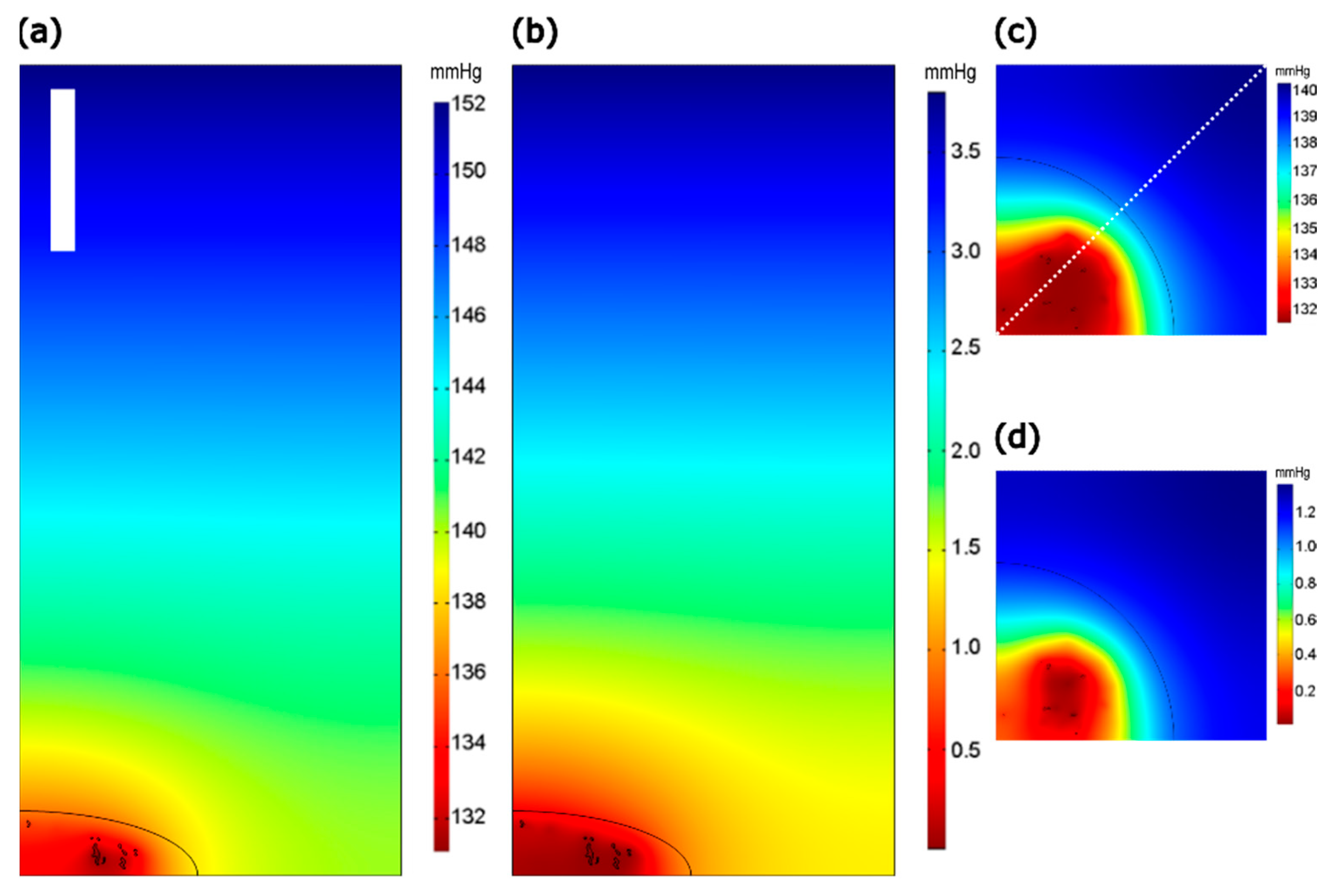
2.2. COMSOL Modeling of the Intracellular pO2 Gradients
3. Discussion
3.1. Effects of the Mitochondrial O2 Consumption and Intracellular pO2 Gradient in z
3.2. The Potential Effects of pH and Refractive Index on the Lifetime Values of Myo-mCherry
3.3. Simplifications of O2 Transport in Media
3.4. Uncertainty in Intracellular O2 Transport Mechanisms
4. Materials and Methods
4.1. FLIM-Based Oxygen Probes
4.2. Cell Transfection with O2 Probes
4.3. Mitochondrial Modulator Treatment
4.4. FLIM Setup
4.5. Fluorescence Microscopy
4.6. FLIM Analysis and Intracellular Mapping of pO2
4.7. Statistical Analysis
4.8. COMSOL Simulation of Intracellular pO2 Gradients
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, D.F.; Rumsey, W.L.; Green, T.J.; Vanderkooi, J.M. The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J. Biol. Chem. 1988, 263, 2712–2718. [Google Scholar] [CrossRef]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A., Jr.; Clark, P.A. Local oxygen gradients near isolated mitochondria. Biophys. J. 1985, 48, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Grinberg, O.Y.; James, P.E.; Swartz, H.M. Are there Significant Gradients of pO2 in Cells. In Oxygen Transport to Tissue XX; Hudetz, A.G., Bruley, D.F., Eds.; Springer: Boston, MA, USA, 1998; pp. 415–423. ISBN 978-1-4615-4863-8. [Google Scholar] [CrossRef]
- Glockner, J.F.; Swartz, H.M.; Pals, M.A. Oxygen gradients in CHO cells: Measurement and characterization by electron spin resonance. J. Cell. Physiol. 1989, 140, 505–511. [Google Scholar] [CrossRef]
- Takahashi, E.; Sato, K.; Endoh, H.; Xu, Z.L.; Doi, K. Direct observation of radial intracellular pO2 gradients in a single cardiomyocyte of the rat. Am. J. Physiol. 1998, 275, H225–H233. [Google Scholar] [CrossRef]
- Katz, I.R.; Wittenberg, J.B.; Wittenberg, B.A. Monoamine oxidase, an intracellular probe of oxygen pressure in isolated cardiac myocytes. J. Biol. Chem. 1984, 259, 7504–7509. [Google Scholar] [CrossRef]
- Jones, D.P. Intracellular diffusion gradients of O2 and ATP. Am. J. Physiol. 1986, 250, C663–C675. [Google Scholar] [CrossRef]
- Kennedy, F.G.; Jones, D.P. Oxygen dependence of mitochondrial function in isolated rat cardiac myocytes. Am. J. Physiol. 1986, 250, C374–C383. [Google Scholar] [CrossRef]
- Robiolio, M.; Rumsey, W.L.; Wilson, D.F. Oxygen diffusion and mitochondrial respiration in neuroblastoma cells. Am. J. Physiol. 1989, 256, C1207–C1213. [Google Scholar] [CrossRef]
- Subczynski, W.K.; Hopwood, L.E.; Hyde, J.S. Is the mammalian cell plasma membrane a barrier to oxygen transport? J. Gen. Physiol. 1992, 100, 69–87. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; Clark, P.A.; Connett, R.J.; Gayeski, T.E.; Honig, C.R. How large is the drop in pO2 between cytosol and mitochondrion? Am. J. Physiol. 1987, 252, C583–C587. [Google Scholar] [CrossRef] [PubMed]
- Hermán, P.; Trübel, H.K.; Hyder, F. A Multiparametric Assessment of Oxygen Efflux from the Brain. J. Cereb. Blood Flow Metab. 2006, 26, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, D.M.; Colburn, T.D.; Craig, J.C.; Hotta, K.; Kano, Y.; Musch, T.I.; Poole, D.C. Skeletal muscle interstitial O2 pressures: Bridging the gap between the capillary and myocyte. Microcirculation 2019, 26, e12497. [Google Scholar] [CrossRef] [PubMed]
- Skattebo, Ø.; Calbet, J.A.L.; Rud, B.; Capelli, C.; Hallén, J. Contribution of oxygen extraction fraction to maximal oxygen uptake in healthy young men. Acta Physiol. Oxf. Engl. 2020, 230, e13486. [Google Scholar] [CrossRef]
- Lesher-Pérez, S.C.; Kim, G.-A.; Kuo, C.-H.; Leung, B.M.; Mong, S.; Kojima, T.; Moraes, C.; Thouless, M.D.; Luker, G.D.; Takayama, S. Dispersible oxygen microsensors map oxygen gradients in three-dimensional cell cultures. Biomater. Sci. 2017, 5, 2106–2113. [Google Scholar] [CrossRef]
- Kutala, V.K.; Parinandi, N.L.; Pandian, R.P.; Kuppusamy, P. Simultaneous measurement of oxygenation in intracellular and extracellular compartments of lung microvascular endothelial cells. Antioxid. Redox Signal. 2004, 6, 597–603. [Google Scholar] [CrossRef]
- Mizukami, K.; Katano, A.; Shiozaki, S.; Yoshihara, T.; Goda, N.; Tobita, S. In vivo O2 imaging in hepatic tissues by phosphorescence lifetime imaging microscopy using Ir(III) complexes as intracellular probes. Sci. Rep. 2020, 10, 21053. [Google Scholar] [CrossRef]
- Kurokawa, H.; Ito, H.; Inoue, M.; Tabata, K.; Sato, Y.; Yamagata, K.; Kizaka-Kondoh, S.; Kadonosono, T.; Yano, S.; Inoue, M.; et al. High resolution imaging of intracellular oxygen concentration by phosphorescence lifetime. Sci. Rep. 2015, 5, 10657. [Google Scholar] [CrossRef] [Green Version]
- James, P.E.; Grinberg, O.Y.; Michaels, G.; Swartz, H.M. Intraphagosomal oxygen in stimulated macrophages. J. Cell. Physiol. 1995, 163, 241–247. [Google Scholar] [CrossRef]
- Rajagopal, V.; Bass, G.; Ghosh, S.; Hunt, H.; Walker, C.; Hanssen, E.; Crampin, E.; Soeller, C. Creating a Structurally Realistic Finite Element Geometric Model of a Cardiomyocyte to Study the Role of Cellular Architecture in Cardiomyocyte Systems Biology. J. Vis. Exp. 2018, 134, 56817. [Google Scholar] [CrossRef]
- Ghosh, S.; Tran, K.; Delbridge, L.M.D.; Hickey, A.J.R.; Hanssen, E.; Crampin, E.J.; Rajagopal, V. Insights on the impact of mitochondrial organisation on bioenergetics in high-resolution computational models of cardiac cell architecture. PLoS Comput. Biol. 2018, 14, e1006640. [Google Scholar] [CrossRef] [Green Version]
- Penjweini, R.; Roarke, B.; Alspaugh, G.; Gevorgyan, A.; Andreoni, A.; Pasut, A.; Sackett, D.L.; Knutson, J.R. Single cell-based fluorescence lifetime imaging of intracellular oxygenation and metabolism. Redox Biol. 2020, 34, 101549. [Google Scholar] [CrossRef] [PubMed]
- Penjweini, R.; Andreoni, A.; Rosales, T.; Kim, J.; Brenner, M.D.; Sackett, D.L.; Chung, J.H.; Knutson, J.R. Intracellular oxygen mapping using a myoglobin-mCherry probe with fluorescence lifetime imaging. J. Biomed. Opt. 2018, 23, 107001. [Google Scholar] [CrossRef]
- Keeley, T.P.; Mann, G.E. Defining Physiological Normoxia for Improved Translation of Cell Physiology to Animal Models and Humans. Physiol. Rev. 2019, 99, 161–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free Radic. Biol. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef]
- Jiang, B.H.; Semenza, G.L.; Bauer, C.; Marti, H.H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996, 271, C1172–C1180. [Google Scholar] [CrossRef] [Green Version]
- Dexter, S.T. Respiratory Rate and Enzyme Activity as Related to the Hardened Condition of Plants. Plant Physiol. 1934, 9, 831–837. [Google Scholar] [CrossRef]
- Hou, H.; Zhao, Y.; Li, C.; Wang, M.; Xu, X.; Jin, Y. Single-cell pH imaging and detection for pH profiling and label-free rapid identification of cancer-cells. Sci. Rep. 2017, 7, 1759. [Google Scholar] [CrossRef] [Green Version]
- Llopis, J.; McCaffery, J.M.; Miyawaki, A.; Farquhar, M.G.; Tsien, R.Y. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 6803–6808. [Google Scholar] [CrossRef]
- Ponsford, A.H.; Ryan, T.A.; Raimondi, A.; Cocucci, E.; Wycislo, S.A.; Frohlich, F.; Swan, L.E.; Stagi, M. Live imaging of intra-lysosome pH in cell lines and primary neuronal culture using a novel genetically encoded biosensor. Autophagy 2021, 17, 1500–1518. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chin, L.K.; Ser, W.; Chen, H.F.; Hsieh, C.M.; Lee, C.H.; Sung, K.B.; Ayi, T.C.; Yap, P.H.; Liedberg, B.; et al. Cell refractive index for cell biology and disease diagnosis: Past, present and future. Lab Chip 2016, 16, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Wong, Y.C.; Gao, F.; Krainc, D. Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson’s disease. Nat. Commun. 2021, 12, 1807. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.; Tozzini, V. Diffusion within the Cytoplasm: A Mesoscale Model of Interacting Macromolecules. Biophys. J. 2014, 107, 2579–2591. [Google Scholar] [CrossRef] [Green Version]
- Keren, K.; Yam, P.T.; Kinkhabwala, A.; Mogilner, A.; Theriot, J.A. Intracellular fluid flow in rapidly moving cells. Nat. Cell Biol. 2009, 11, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.; Scheiner, A.; Cunningham, J.; Gatenby, R. Cytoplasmic convection currents and intracellular temperature gradients. PLoS Comput. Biol. 2019, 15, e1007372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, D.C.; Wenning, L.A.; Miller, W.M.; Papoutsakis, E.T. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I. Krogh’s model. Biophys. J. 2001, 81, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Chow, D.C.; Wenning, L.A.; Miller, W.M.; Papoutsakis, E.T. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys. J. 2001, 81, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Fischkoff, S.; Vanderkooi, J.M. Oxygen diffusion in biological and artificial membranes determined by the fluorochrome pyrene. J. Gen. Physiol. 1975, 65, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Vadapalli, A.; Pittman, R.N.; Popel, A.S. Estimating oxygen transport resistance of the microvascular wall. Am. J. Physiol.-Heart Circ. Physiol. 2000, 279, H657–H671. [Google Scholar] [CrossRef]
- Carrier, R.L.; Rupnick, M.; Langer, R.; Schoen, F.J.; Freed, L.E.; Vunjak-Novakovic, G. Perfusion Improves Tissue Architecture of Engineered Cardiac Muscle. Tissue Eng. 2002, 8, 175–188. [Google Scholar] [CrossRef]
- Goldstick, T.K.; Ciuryla, V.T.; Zuckerman, L. Diffusion of oxygen in plasma and blood. Adv. Exp. Med. Biol. 1976, 75, 183–190. [Google Scholar] [PubMed]
- Wagner, B.A.; Venkataraman, S.; Buettner, G.R. The rate of oxygen utilization by cells. Free Radic. Biol. Med. 2011, 51, 700–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, A.; Shi, R.; Luciani, D.S. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic β-cells. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E87–E101. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, A.; Beichel, R.; Kalpathy-Cramer, J.; Finet, J.; Fillion-Robin, J.C.; Pujol, S.; Bauer, C.; Jennings, D.; Fennessy, F.; Sonka, M.; et al. 3D Slicer as an image computing platform for the Quantitative Imaging Network. Magn. Reson. Imaging 2012, 30, 1323–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattei, G.; Giusti, S.; Ahluwalia, A. Design Criteria for Generating Physiologically Relevant In Vitro Models in Bioreactors. Processes 2014, 2, 548–569. [Google Scholar] [CrossRef] [Green Version]
- Pias, S.C. How does oxygen diffuse from capillaries to tissue mitochondria? Barriers and pathways. J. Physiol. 2021, 599, 1769–1782. [Google Scholar] [CrossRef] [PubMed]
- Pannala, V.R.; Camara, A.K.S.; Dash, R.K. Modeling the detailed kinetics of mitochondrial cytochrome c oxidase: Catalytic mechanism and nitric oxide inhibition. J. Appl. Physiol. 2016, 121, 1196–1207. [Google Scholar] [CrossRef] [Green Version]
- Scandurra, F.M.; Gnaiger, E. Cell respiration under hypoxia: Facts and artefacts in mitochondrial oxygen kinetics. Adv. Exp. Med. Biol. 2010, 662, 7–25. [Google Scholar]
- Suarez, R.K.; Lighton, J.R.; Brown, G.S.; Mathieu-Costello, O. Mitochondrial respiration in hummingbird flight muscles. Proc. Natl. Acad. Sci. USA 1991, 88, 4870–4873. [Google Scholar] [CrossRef] [Green Version]
- Suarez, R.K. Upper limits to mass-specific metabolic rates. Annu. Rev. Physiol. 1996, 58, 583–605. [Google Scholar] [CrossRef]
- Syromyatnikov, M.Y.; Gureev, A.P.; Vitkalova, I.Y.; Starkov, A.A.; Popov, V.N. Unique features of flight muscles mitochondria of honey bees (Apis mellifera L.). Arch. Insect Biochem. Physiol. 2019, 102, e21595. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle. Int. J. Biochem. Cell Biol. 2009, 41, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.A.; Lundby, C. Contextualizing the biological relevance of standardized high-resolution respirometry to assess mitochondrial function in permeabilized human skeletal muscle. Acta Physiol. Oxf. Engl. 2021, 231, e13625. [Google Scholar] [CrossRef] [PubMed]
- Himmelblau, D.M. Diffusion of Dissolved Gases in Liquids. Chem. Rev. 1964, 64, 527–550. [Google Scholar] [CrossRef]
- Longeville, S.; Stingaciu, L.-R. Hemoglobin diffusion and the dynamics of oxygen capture by red blood cells. Sci. Rep. 2017, 7, 10448. [Google Scholar] [CrossRef] [Green Version]
- Moschandreou, T.E.; Ellis, C.G.; Goldman, D. Influence of tissue metabolism and capillary oxygen supply on arteriolar oxygen transport: A computational model. Math. Biosci. 2011, 232, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Radisic, M.; Deen, W.; Langer, R.; Vunjak-Novakovic, G. Mathematical model of oxygen distribution in engineered cardiac tissue with parallel channel array perfused with culture medium containing oxygen carriers. Am. J. Physiol.-Heart Circ. Physiol. 2005, 288, H1278–H1289. [Google Scholar] [CrossRef]
- Hamon, M.; Hanada, S.; Fujii, T.; Sakai, Y. Direct oxygen supply with polydimethylsiloxane (PDMS) membranes induces a spontaneous organization of thick heterogeneous liver tissues from rat fetal liver cells in vitro. Cell Transpl. 2012, 21, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Morsiani, E.; Brogli, M.; Galavotti, D.; Bellini, T.; Ricci, D.; Pazzi, P.; Puviani, A.C. Long-Term Expression of Highly Differentiated Functions by Isolated Porcine Hepatocytes Perfused in a Radial-Flow Bioreactor. Artif. Organs 2001, 25, 740–748. [Google Scholar] [CrossRef]
- Melendez-Ferro, M.; Rice, M.W.; Roberts, R.C.; Perez-Costas, E. An accurate method for the quantification of cytochrome C oxidase in tissue sections. J. Neurosci. Methods 2013, 214, 156–162. [Google Scholar] [CrossRef]
- Buchwald, P. FEM-based oxygen consumption and cell viability models for avascular pancreatic islets. Biol. Med.. Model 2009, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, R.K. Energy metabolism during insect flight: Biochemical design and physiological performance. Physiol. Biochem. Zool. 2000, 73, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Casey, T.M.; Arthur, P.G. Hibernation in Noncontracting Mammalian Cardiomyocytes. Circulation 2000, 102, 3124–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.-A.; Palsson, B.O. Determination of specific oxygen uptake rates in human hematopoietic cultures and implications for bioreactor design. Ann. Biomed. Eng. 1996, 24, 373–381. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OxyLite Pro | |||
|---|---|---|---|
| τmax | K | R2 | |
| Mitochondria | 1.15 ± 0.03 | 6.889 ± 2.819 | 0.99 |
| Cytosol | 1.17 ± 0.04 | 6.852 ± 3.498 | 0.98 |
| Rot/Antim | 1.29 ± 0.03 | 6.764 ± 1.577 | 0.98 |
| O2. (%) | Spacing (µm) | Lower Boundary | Mean Mitochondrial pO2 (mmHg) | Mean Cytoplasmic pO2 (mmHg) | Difference (mmHg) |
|---|---|---|---|---|---|
| 0.5 | 70 | Glass | 0.01 | 0.23 | 0.21 |
| 5 | 4.63 | 7.09 | 2.46 | ||
| 10 | 42.1 | 44.6 | 2.50 | ||
| 20 | 118.1 | 120.6 | 2.50 | ||
| 0.5 | 100 | Glass | 0.028 | 0.428 | 0.4 |
| 5 | 19.0 | 21.4 | 2.4 | ||
| 10 | 56.9 | 59.4 | 2.5 | ||
| 20 | 132.9 | 135.4 | 2.5 | ||
| 0.5 | 200 | Glass | 0.064 | 0.764 | 0.7 |
| 5 | 25.7 | 28.3 | 2.6 | ||
| 10 | 63.6 | 66.3 | 2.7 | ||
| 20 | 139.6 | 142.3 | 2.7 | ||
| 0.5 | 70 | Ibidi Polymer | 0.01 | 0.27 | 0.25 |
| 5 | 5.22 | 7.68 | 2.46 | ||
| 10 | 42.6 | 45.1 | 2.49 | ||
| 20 | 118.2 | 120.7 | 2.5 | ||
| 0.5 | 100 | Ibidi Polymer | 0.036 | 0.508 | 0.5 |
| 5 | 19.6 | 22.1 | 2.5 | ||
| 10 | 57.4 | 59.8 | 2.5 | ||
| 20 | 133.0 | 135.5 | 2.5 | ||
| 0.5 | 200 | Ibidi Polymer | 0.087 | 0.914 | 0.8 |
| 5 | 26.2 | 28.8 | 2.6 | ||
| 10 | 64.0 | 66.7 | 2.7 | ||
| 20 | 139.7 | 142.3 | 2.7 | ||
| 0.5 | 70 | 200 µm PDMS | 0.38 | 1.27 | 0.89 |
| 5 | 33.2 | 34.5 | 1.31 | ||
| 10 | 71.2 | 72.5 | 1.32 | ||
| 20 | 147.2 | 148.5 | 1.32 | ||
| 0.5 | 100 | 200 µm PDMS | 0.779 | 1.87 | 1.1 |
| 5 | 34.5 | 35.8 | 1.3 | ||
| 10 | 72.6 | 73.8 | 1.3 | ||
| 20 | 148.5 | 149.8 | 1.3 | ||
| 0.5 | 200 | 200 µm PDMS | 1.08 | 2.30 | 1.2 |
| 5 | 35.0 | 36.4 | 1.4 | ||
| 10 | 73.0 | 74.4 | 1.4 | ||
| 20 | 149.0 | 150.4 | 1.4 |
| Description | Symbol [Units] | Element | Value | References |
|---|---|---|---|---|
| O2 diffusivity ‘consensus’ | D [cm2/s] | Medium, | 1.9 × 10−5 | [16,26,56,57,58] |
| intracellular | 0.9 × 10−5 | [37,38,39,40,41,42] | ||
| PDMS | 5 × 10−5 | [59] | ||
| O2 solubility | S [mol/cm3/mmHg] | Medium, intracellular | 1.29 × 10−3 | [16,38,60,61] |
| PDMS | 1.06 × 10−2 | [59] | ||
| O2 permeability | P [cm/s] | Membrane | 42 | [11] |
| PDMS | 8 | [62] | ||
| Slide mass transfer coefficient | k [mol/cm2/s/bar O2] | Slide | 7.19 × 10−12 | Ibidi manufacturer |
| Maximum O2 consumption rate | Vmax [mol/m3/s] | Mitochondria | 10 | [37,38,43,50,63] |
| [O2] at half-maximal rate | Km [mol/m3] | Mitochondria | 1 × 10−4 | [26,58,64,65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedlack, A.J.H.; Penjweini, R.; Link, K.A.; Brown, A.; Kim, J.; Park, S.-J.; Chung, J.H.; Morgan, N.Y.; Knutson, J.R. Computational Modeling and Imaging of the Intracellular Oxygen Gradient. Int. J. Mol. Sci. 2022, 23, 12597. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012597
Sedlack AJH, Penjweini R, Link KA, Brown A, Kim J, Park S-J, Chung JH, Morgan NY, Knutson JR. Computational Modeling and Imaging of the Intracellular Oxygen Gradient. International Journal of Molecular Sciences. 2022; 23(20):12597. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012597
Chicago/Turabian StyleSedlack, Andrew J. H., Rozhin Penjweini, Katie A. Link, Alexandra Brown, Jeonghan Kim, Sung-Jun Park, Jay H. Chung, Nicole Y. Morgan, and Jay R. Knutson. 2022. "Computational Modeling and Imaging of the Intracellular Oxygen Gradient" International Journal of Molecular Sciences 23, no. 20: 12597. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232012597