Inherited Retinal Diseases

Ruth and Bruce Rappaport Faculty of Medicine, Technion-Israel Institute of Technology, Haifa 3109601, Israel
Int. J. Mol. Sci. 2022, 23(21), 13467; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232113467
Submission received: 30 October 2022
/
Accepted: 2 November 2022
/
Published: 3 November 2022
(This article belongs to the Special Issue Inherited Retinal Diseases)
{kind=link}
Inherited retinal diseases (IRDs) are a clinically and genetically heterogeneous group of diseases that cause vision loss due to abnormal development or due to the dysfunction or degeneration of the photoreceptors or the retinal pigment epithelium. They have a prevalence of approximately 1:1380 individuals, with 5.5 million people expected to be affected worldwide. The most common form of IRD is Retinitis Pigmentosa. Other forms include cone/cone–rod degeneration, Leber congenital amaurosis, inherited macular dystrophies, among others [1]. While in most IRD cases, the disease only involves ophthalmic manifestations (nonsyndromic), over 70 forms of syndromic IRDs have been described [2]. The most common form of syndromic IRD is Usher syndrome (see review by Fuster-Garcia et al., in this Special Issue) [3]). The aim of this Special Issue was to focus on the major challenges in the IRD field, including phenotypic characterization, molecular diagnosis, functional studies, and therapy.
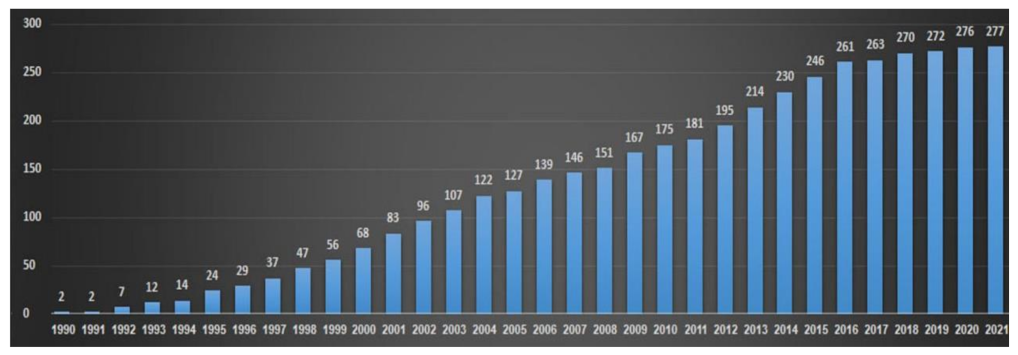
IRD is the most genetically heterogeneous group of disorders in humans. It can be inherited as autosomal recessive, autosomal dominant, or X-linked. Mitochondrial and digenic patterns of inheritance have also been described. Since the identification of the first two IRD-causative genes back in 1990, RHO and CHM [4,5], an average of nine newly identified causative genes have been identified every year between the years 1990 and 2021, and to date, at least 280 genes have been implicated in IRD (RetNet at https://sph.uth.edu/Retnet/, accessed on 30 October 2022). However, this discovery phase has now reached a plateau (Figure 1), indicating that while some rare novel IRD genes may exist, the vast majority of IRD-causative genes have already been discovered. Nevertheless, the contribution of each of these genes to the overall prevalence of the disease is relatively small, and 65% of identified pathogenic variants are unique to a single individual [1]. Moreover, despite the wide application of Next-Generation Sequencing (NGS) on large IRD cohorts, reported mutation identification rates worldwide range between 50 and 76%. This is true not only for the cohorts analyzed by whole exome sequencing or targeted NGS (i.e., gene panels), but also for cohorts analyzed by whole genome sequencing [6,7,8]. Taken together, the conclusion is that most of the missing mutations are located in known IRD genes and are not yet identified, either due to technical limitations or to misinterpretation. The significant proportion of cases that remain unsolved (up to 30%) poses a challenge for both clinical and research aspects. Accurate, sensitive, and cost-effective genetic analysis of IRD patients therefore requires the combined development of efficient sequencing and bioinformatics tools as well as thoughtful analysis workflows. Moreover, functional evaluation is required to prove the pathogenicity of some of the identified variants. Excellent examples for such developments are provided and reviewed in this Special Issue [9,10,11,12,13,14].
In recent years, the IRD field has undergone dramatic changes, mainly due to the development of novel therapeutic strategies that are either non-gene-based (stem cell therapy and retinal implants), gene-based (gene therapy and targeted pharmacological agents), or mutation-based (translational read-through inducing drugs, antisense oligonucleotides; The latter is used in the study by Tomkiewicz et al. [15]). A large number of clinical trials targeting various IRD-related genes and mutations are currently ongoing. Comprehensive studies on the clinical spectrum and natural history associated with various IRD-causative genes and mutations are greatly important not only to define prognosis, but also to identify clinical trial endpoints [14,16,17,18,19,20,21,22,23].
Finally, the biological function and disease etiology of many of the genes associated with IRD are not fully understood. Studies addressing these questions while using various experimental systems (such as the ones by Aísa-Marín et al., Koch et al., and Remez et al. [24,25,26]) provide significant insights into mechanisms of normal retinal function and the types of cellular defects that lead to retinal degeneration. These findings will help in the prevention and development of new therapeutic strategies for retinal degeneration.
Funding
This research received no external funding.
Acknowledgments
I thank all of the authors who contributed to this Special Issue and Susanne Roosing, who served as a Co-Editor of the Special Issue.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| IRD | Inherited Retinal Diseases |
| NGS | Next-Generation Sequencing |
References
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 2021, 89, 101029. [Google Scholar] [CrossRef] [PubMed]
- Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics 2020, 10, 779. [Google Scholar] [CrossRef]
- Fuster-García, C.; García-Bohórquez, B.; Rodríguez-Muñoz, A.; Aller, E.; Jaijo, T.; Millán, J.M.; García-García, G. Usher Syndrome: Genetics of a Human Ciliopathy. Int. J. Mol. Sci. 2021, 22, 6723. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.; van de Pol, D.J.; van Kerkhoff, L.P.; Wieringa, B.; Ropers, H.H. Cloning of a gene that is rearranged in patients with choroideraemia. Nature 1990, 347, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; McGee, T.L.; Hahn, L.B.; Cowley, G.S.; Olsson, J.E.; Reichel, E.; Sandberg, M.A.; Berson, E.L. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 1990, 323, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Del Pozo-Valero, M.; Riveiro-Alvarez, R.; Martin-Merida, I.; Blanco-Kelly, F.; Swafiri, S.; Lorda-Sanchez, I.; Trujillo-Tiebas, M.J.; Carreno, E.; Jimenez-Rolando, B.; Garcia-Sandoval, B.; et al. Impact of Next Generation Sequencing in Unraveling the Genetics of 1036 Spanish Families With Inherited Macular Dystrophies. Investig. Opthalmology Vis. Sci. 2022, 63, 11. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Atac, D.; Mohn, L.; Feil, S.; Maggi, K.; Haenni, D.; Seebauer, B.; Koller, S.; Berger, W. Functional Characterization of an In-Frame Deletion in the Basic Domain of the Retinal Transcription Factor ATOH7. Int. J. Mol. Sci. 2022, 23, 1053. [Google Scholar] [CrossRef]
- de Bruijn, S.E.; Fadaie, Z.; Cremers, F.P.M.; Kremer, H.; Roosing, S. The Impact of Modern Technologies on Molecular Diagnostic Success Rates, with a Focus on Inherited Retinal Dystrophy and Hearing Loss. Int. J. Mol. Sci. 2021, 22, 2943. [Google Scholar] [CrossRef]
- Kortüm, F.; Kieninger, S.; Mazzola, P.; Kohl, S.; Wissinger, B.; Prokisch, H.; Stingl, K.; Weisschuh, N. X-Linked Retinitis Pigmentosa Caused by Non-Canonical Splice Site Variants in RPGR. Int. J. Mol. Sci. 2021, 22, 850. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sánchez, M.; Bravo-Gil, N.; González-del Pozo, M.; Méndez-Vidal, C.; Fernández-Suárez, E.; Rodríguez-de la Rúa, E.; Borrego, S.; Antiñolo, G. A Multi-Strategy Sequencing Workflow in Inherited Retinal Dystrophies: Routine Diagnosis, Addressing Unsolved Cases and Candidate Genes Identification. Int. J. Mol. Sci. 2020, 21, 9355. [Google Scholar] [CrossRef] [PubMed]
- Reurink, J.; Dockery, A.; Oziębło, D.; Farrar, G.J.; Ołdak, M.; ten Brink, J.B.; Bergen, A.A.; Rinne, T.; Yntema, H.G.; Pennings, R.J.E.; et al. Molecular Inversion Probe-Based Sequencing of USH2A Exons and Splice Sites as a Cost-Effective Screening Tool in USH2 and arRP Cases. Int. J. Mol. Sci. 2021, 22, 6419. [Google Scholar] [CrossRef]
- Weisschuh, N.; Mazzola, P.; Bertrand, M.; Haack, T.B.; Wissinger, B.; Kohl, S.; Stingl, K. Clinical Characteristics of POC1B-Associated Retinopathy and Assignment of Pathogenicity to Novel Deep Intronic and Non-Canonical Splice Site Variants. Int. J. Mol. Sci. 2021, 22, 5396. [Google Scholar] [CrossRef] [PubMed]
- Tomkiewicz, T.Z.; Suárez-Herrera, N.; Cremers, F.P.M.; Collin, R.W.J.; Garanto, A. Antisense Oligonucleotide-Based Rescue of Aberrant Splicing Defects Caused by 15 Pathogenic Variants in ABCA4. Int. J. Mol. Sci. 2021, 22, 4621. [Google Scholar] [CrossRef]
- Abad-Morales, V.; Wert, A.; Gómez, M.Á.R.; Navarro, R.; Pomares, E. New Insights on the Genetic Basis Underlying SHILCA Syndrome: Characterization of the NMNAT1 Pathological Alterations Due to Compound Heterozygous Mutations and Identification of a Novel Alternative Isoform. Int. J. Mol. Sci. 2021, 22, 2262. [Google Scholar] [CrossRef]
- Hufendiek, K.; Hufendiek, K.; Jägle, H.; Stöhr, H.; Book, M.; Spital, G.; Rustambayova, G.; Framme, C.; Weber, B.H.F.; Renner, A.B.; et al. Clinical Heterogeneity in Autosomal Recessive Bestrophinopathy with Biallelic Mutations in the BEST1 Gene. Int. J. Mol. Sci. 2020, 21, 9353. [Google Scholar] [CrossRef]
- Iarossi, G.; Marino, V.; Maltese, P.E.; Colombo, L.; D’Esposito, F.; Manara, E.; Dhuli, K.; Modarelli, A.M.; Cennamo, G.; Magli, A.; et al. Expanding the Clinical and Genetic Spectrum of RAB28-Related Cone-Rod Dystrophy: Pathogenicity of Novel Variants in Italian Families. Int. J. Mol. Sci. 2021, 22, 381. [Google Scholar] [CrossRef]
- Inaba, A.; Maeda, A.; Yoshida, A.; Kawai, K.; Hirami, Y.; Kurimoto, Y.; Kosugi, S.; Takahashi, M. Truncating Variants Contribute to Hearing Loss and Severe Retinopathy in USH2A-Associated Retinitis Pigmentosa in Japanese Patients. Int. J. Mol. Sci. 2020, 21, 7817. [Google Scholar] [CrossRef]
- Jackson, D.J.; Dubis, A.M.; Moosajee, M. The Natural History of CNGB1-Related Retinopathy: A Longitudinal Phenotypic Analysis. Int. J. Mol. Sci. 2022, 23, 6785. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Sumaroka, A.; Roman, A.J.; Wu, V.; Swider, M.; Sheplock, R.; Krishnan, A.K.; Garafalo, A.V. Leber Congenital Amaurosis Due to GUCY2D Mutations: Longitudinal Analysis of Retinal Structure and Visual Function. Int. J. Mol. Sci. 2021, 22, 2031. [Google Scholar] [CrossRef] [PubMed]
- Kellner, U.; Weisschuh, N.; Weinitz, S.; Farmand, G.; Deutsch, S.; Kortüm, F.; Mazzola, P.; Schäferhoff, K.; Marino, V.; Dell’Orco, D. Autosomal Dominant Gyrate Atrophy-Like Choroidal Dystrophy Revisited: 45 Years Follow-Up and Association with a Novel C1QTNF5 Missense Variant. Int. J. Mol. Sci. 2021, 22, 2089. [Google Scholar] [CrossRef] [PubMed]
- Kuehlewein, L.; Zobor, D.; Stingl, K.; Kempf, M.; Nasser, F.; Bernd, A.; Biskup, S.; Cremers, F.P.M.; Khan, M.I.; Mazzola, P.; et al. Clinical Phenotype of PDE6B-Associated Retinitis Pigmentosa. Int. J. Mol. Sci. 2021, 22, 2374. [Google Scholar] [CrossRef]
- Aísa-Marín, I.; García-Arroyo, R.; Mirra, S.; Marfany, G. The Alter Retina: Alternative Splicing of Retinal Genes in Health and Disease. Int. J. Mol. Sci. 2021, 22, 1855. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Scheel, C.; Ma, H.; Yang, F.; Stadlmeier, M.; Glück, A.F.; Murenu, E.; Traube, F.R.; Carell, T.; Biel, M.; et al. The cGMP-Dependent Protein Kinase 2 Contributes to Cone Photoreceptor Degeneration in the Cnga3-Deficient Mouse Model of Achromatopsia. Int. J. Mol. Sci. 2021, 22, 52. [Google Scholar] [CrossRef]
- Remez, L.; Cohen, B.; Nevet, M.J.; Rizel, L.; Ben-Yosef, T. TULP1 and TUB Are Required for Specific Localization of PRCD to Photoreceptor Outer Segments. Int. J. Mol. Sci. 2020, 21, 8677. [Google Scholar] [CrossRef]
Figure 1.
IRD-causative gene identification over the years. The chart shows the total number of known IRD-causative genes each year between 1990 and 2021.
Figure 1.
IRD-causative gene identification over the years. The chart shows the total number of known IRD-causative genes each year between 1990 and 2021.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ben-Yosef, T. Inherited Retinal Diseases. Int. J. Mol. Sci. 2022, 23, 13467. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232113467
AMA Style
Ben-Yosef T. Inherited Retinal Diseases. International Journal of Molecular Sciences. 2022; 23(21):13467. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232113467
Chicago/Turabian StyleBen-Yosef, Tamar. 2022. "Inherited Retinal Diseases" International Journal of Molecular Sciences 23, no. 21: 13467. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232113467
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.