
Reactive Microgliosis in Sepsis-Associated and Acute Hepatic Encephalopathies: An Ultrastructural Study


Abstract
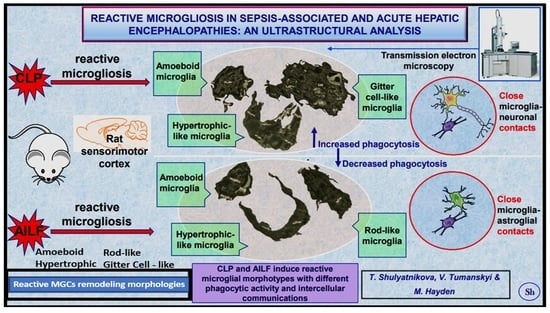
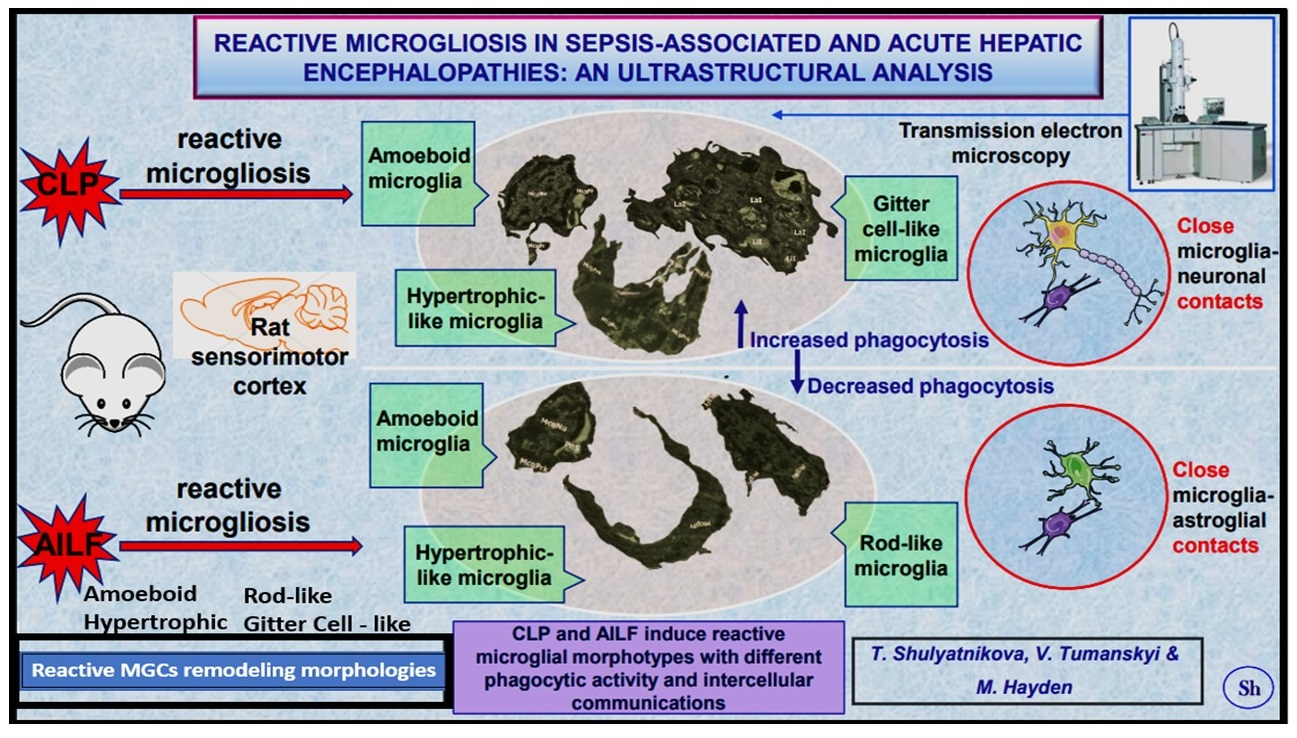
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. General Ultrastructural Histopathology of the CLP and AILF Brain Cortex
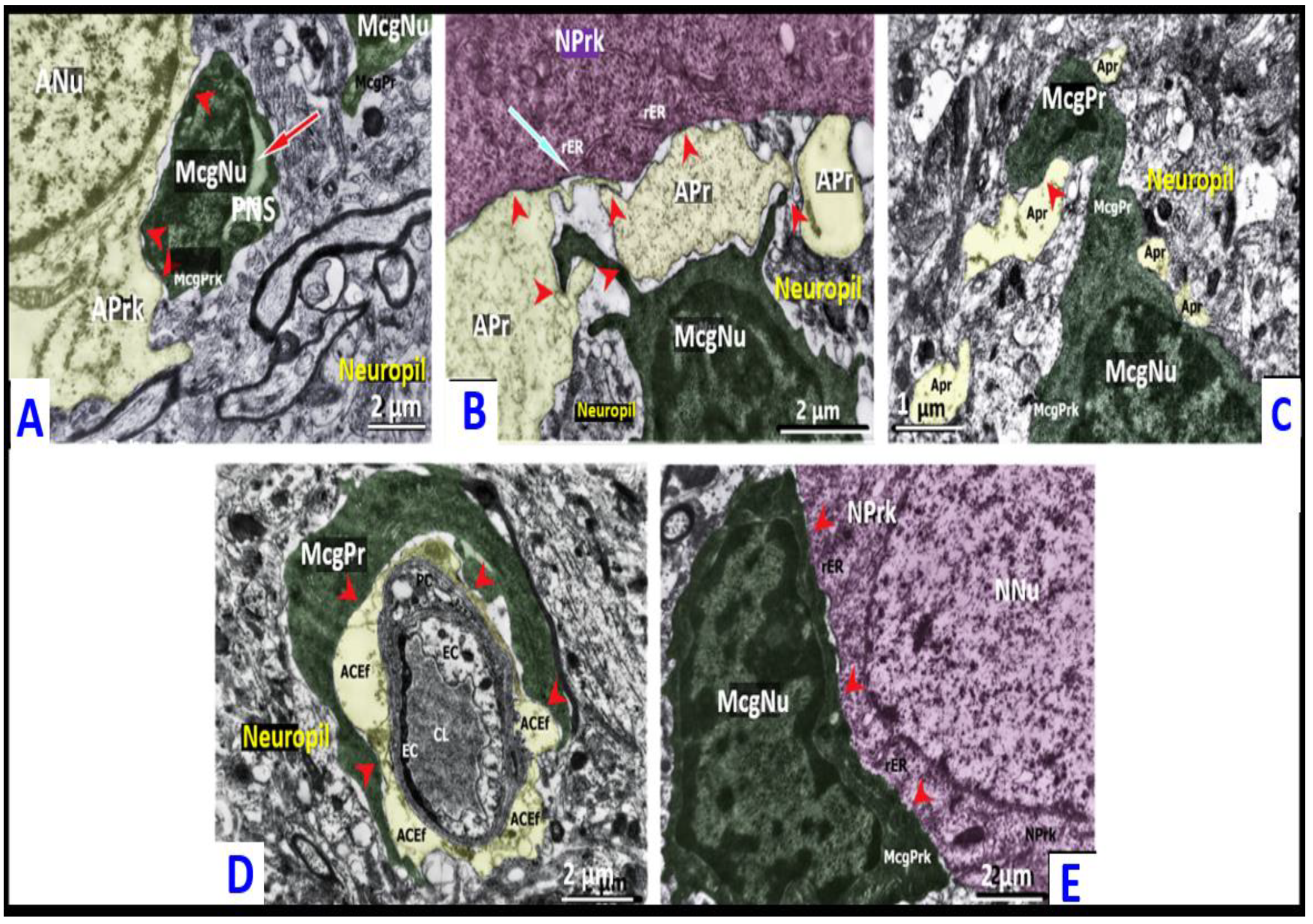
2.1.1. CLP Model
2.1.2. AILF Model
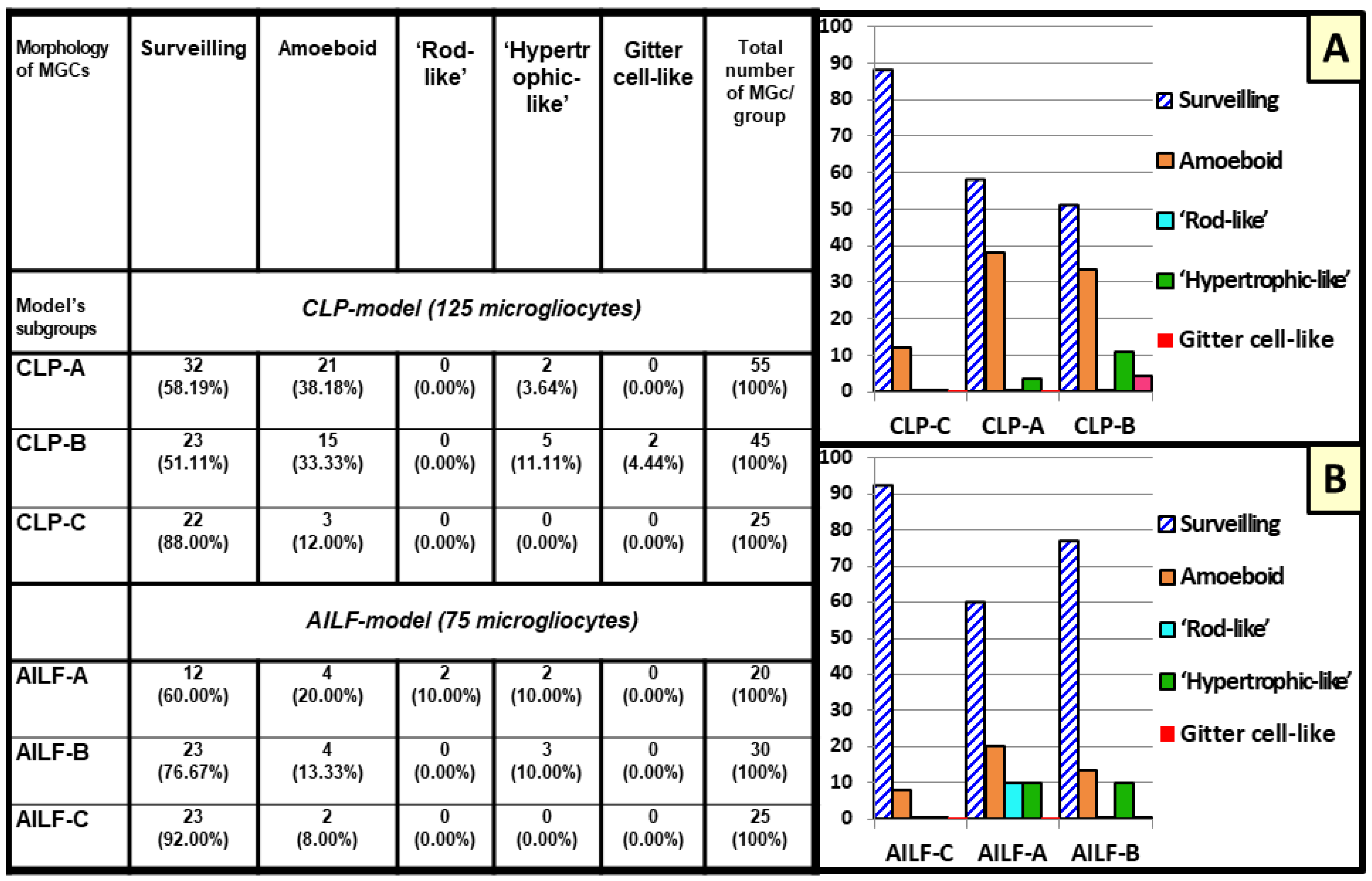
2.2. Microglial Morphotypes in CLP and AILF Models
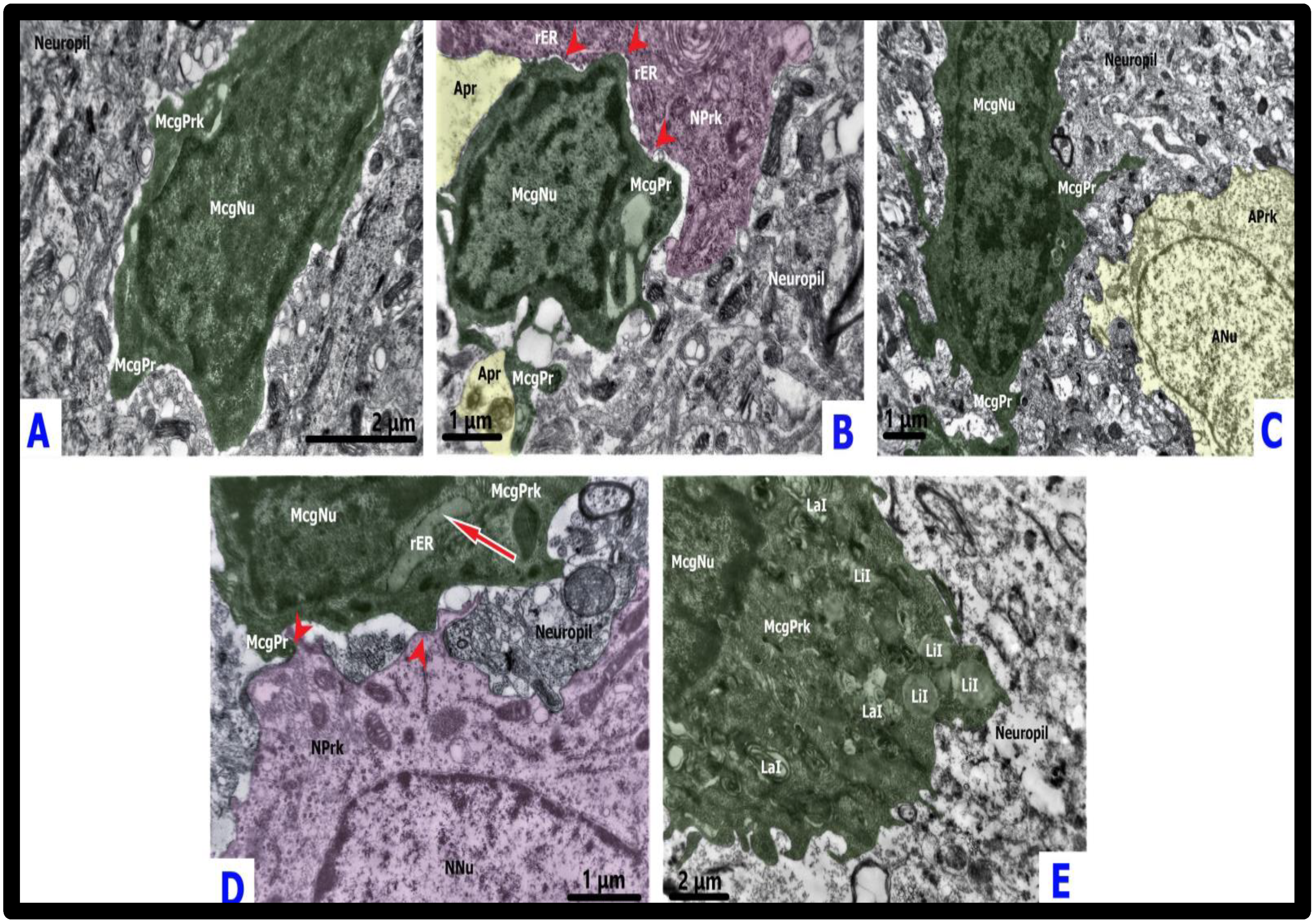
2.2.1. Surveilling Microglia
2.2.2. Amoeboid Microglia
2.2.3. Rod-like Microglia (Stäbchellen)
2.2.4. Hypertrophic-like Microglia
2.2.5. Gitter Cell-like Microglia (Gitterzellen)
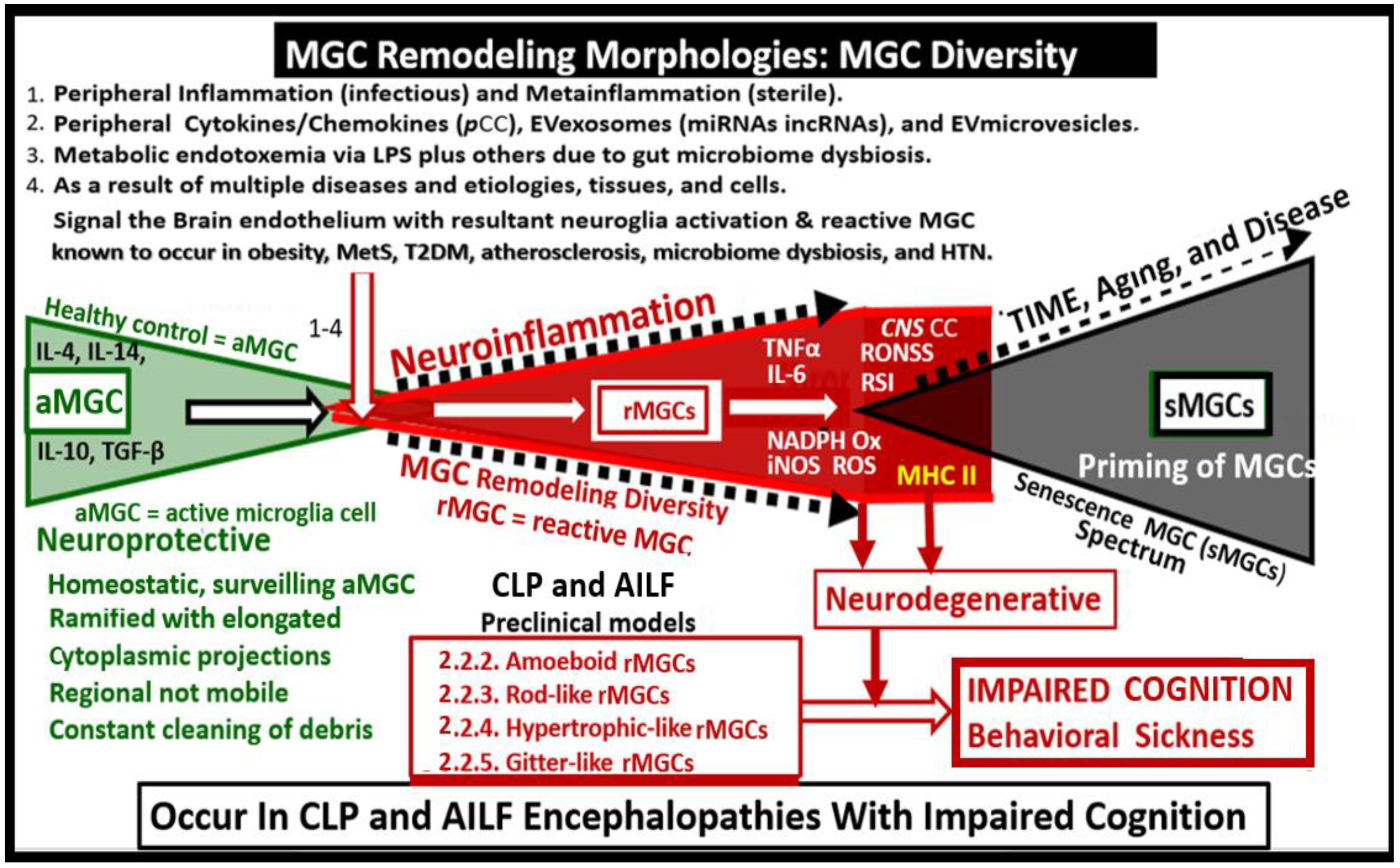
2.3. Reactive Microglia in Sepsis-Associated Encephalopathy
2.4. Reactive Microglia in Acute Hepatic Encephalopathy
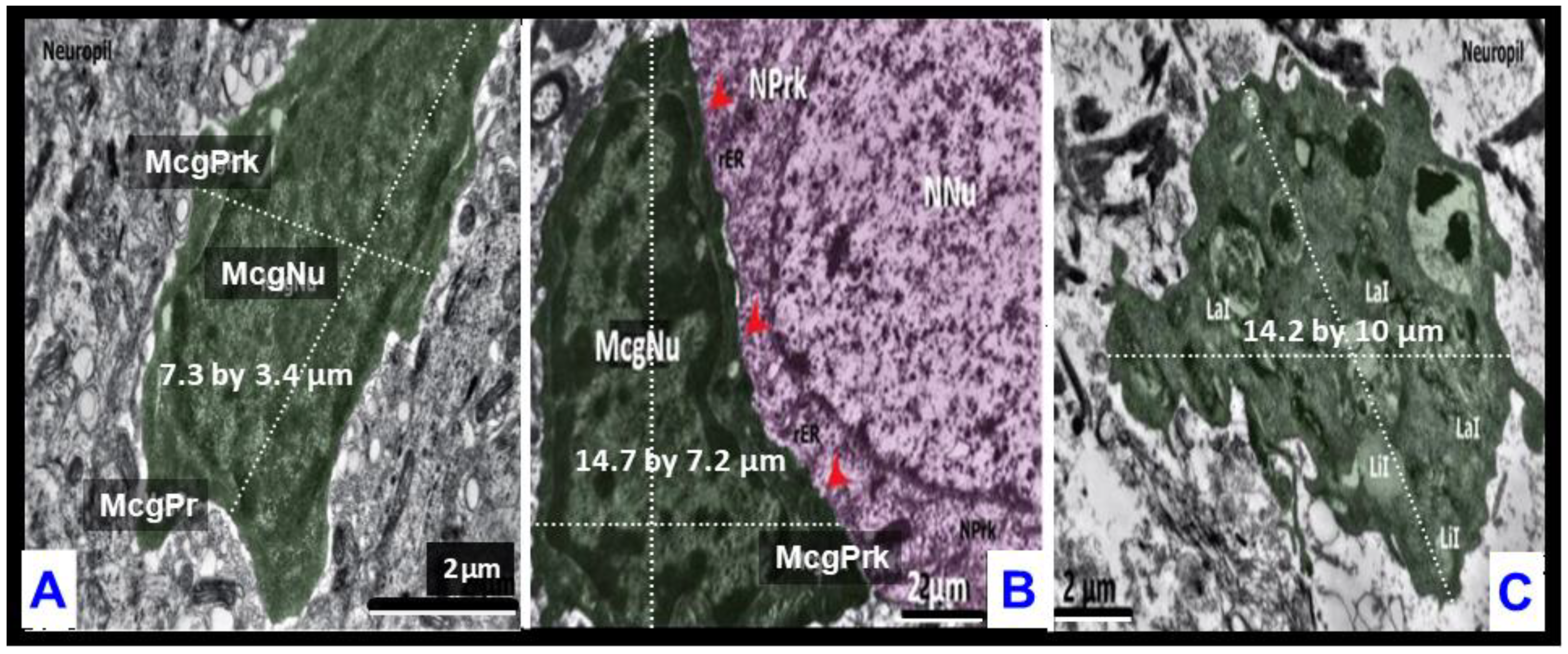
2.5. Hypertrophic-like and Gitter Cell-like Microglia Are Larger Than Surveilling Control Microgliocytes
3. Discussion
3.1. Microglial Reactivity in SAE
3.2. Microglial Reactivity in Acute Hepatic Encephalopathy (AHE)
3.3. Dark Microglia (dMG) as Associated with CLP and AILF Rat Models of Sepsis and Liver Failure-Induced Encephalopathies
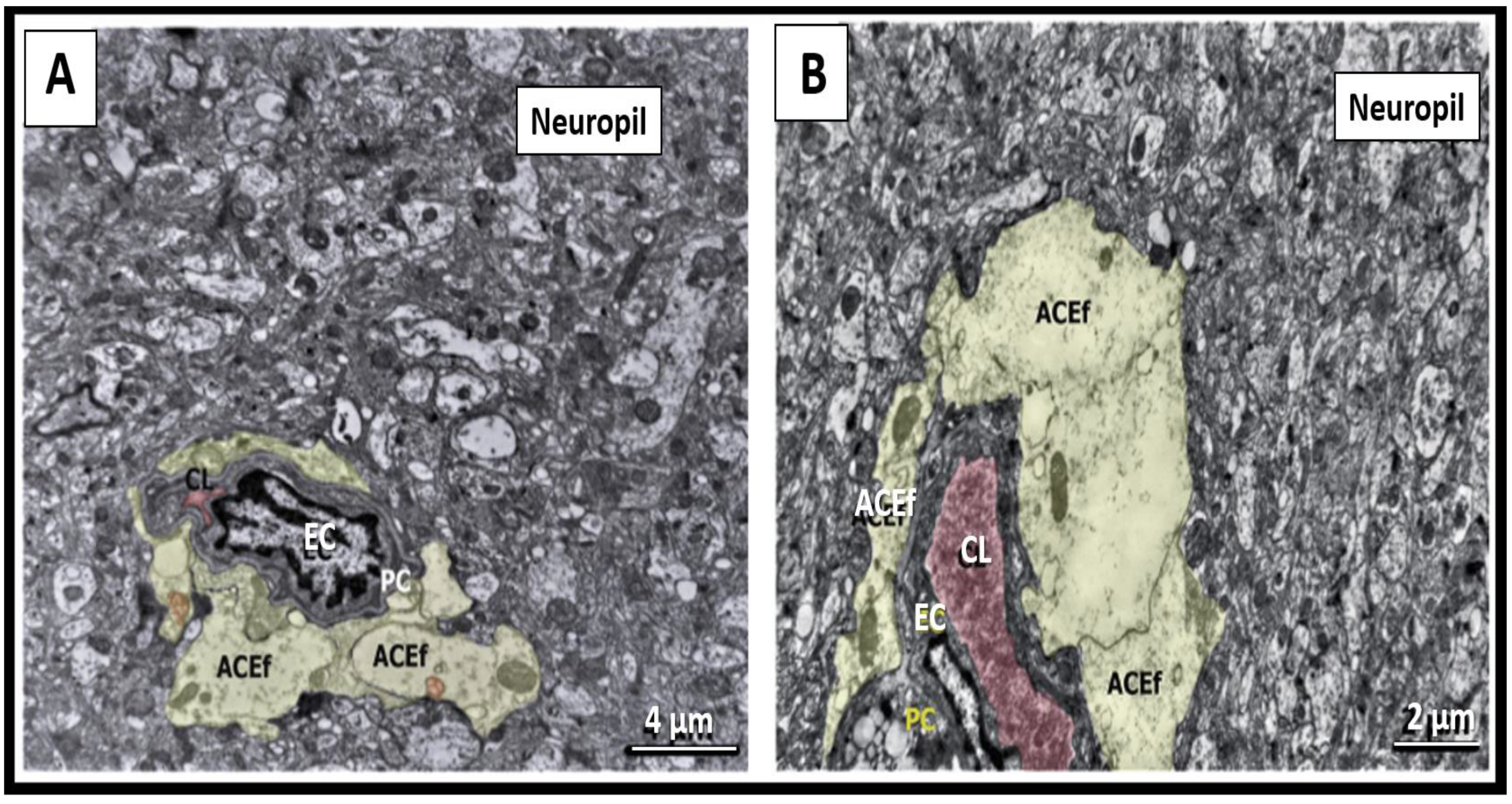
3.4. Peripheral Inflammation Signals the Brain Endothelial Cell(s) (BECs) via an Organ–Brain Axis to Result in Reactive Microglia and Neuroinflammation
3.5. Limitations to This Ultrastructural TEM Study
4. Materials and Methods
4.1. Animals
4.2. Sepsis-Associated Encephalopathy Model
4.3. Acute Hepatic Encephalopathy Model
4.4. Tissue Collection and Preparation for Transmission Electron Microscopy (TEM)
4.5. Ultrastructural and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gao, Q.; Hernandes, M.S. Sepsis-Associated Encephalopathy and Blood-Brain Barrier Dysfunction. Inflammation 2021, 44, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Jain, L.; Sharma, B.C.; Sharma, P.; Srivastava, S.; Agrawal, A.; Sarin, S.K. Serum endotoxin and inflammatory mediators in patients with cirrhosis and hepatic encephalopathy. Dig. Liver Dis. 2012, 44, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Shulyatnikova, T.V.; Shavrin, V.A. Modern view on hepatic encephalopathy: Basic terms and concepts of pathogenesis. Pathologia 2017, 14, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Mazeraud, A.; Righy, C.; Bouchereau, E.; Benghanem, S.; Bozza, F.A.; Sharshar, T. Septic-Associated Encephalopathy: A Comprehensive Review. Neurotherapeutics 2020, 17, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Fogel, W.A.; Andrzejewski, W.; Maśliński, C. Neurotransmitters in hepatic encephalopathy. Acta Neurobiol. Exp. (Wars) 1990, 50, 281–293. [Google Scholar]
- Mizock, B.A.; Sabelli, H.C.; Dubin, A.; Javaid, J.I.; Poulos, A.; Rackow, E.C. Septic encephalopathy. Evidence for altered phenylalanine metabolism and comparison with hepatic encephalopathy. Arch. Intern. Med. 1990, 150, 443–449. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- De Biase, L.M.; Bonci, A. Region-Specific Phenotypes of Microglia: The Role of Local Regulatory Cues. Neuroscientist 2019, 25, 314–333. [Google Scholar] [CrossRef]
- Lee, J.; Hamanaka, G.; Lo, E.H.; Arai, K. Heterogeneity of microglia and their differential roles in white matter pathology. CNS Neurosci. Ther. 2019, 25, 1290–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.-È.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef] [PubMed]
- Michels, M.; Sonai, B.; Dal-Pizzol, F. Polarization of microglia and its role in bacterial sepsis. J. Neuroimmunol. 2017, 303, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Wang, Y.; Stetler, A.R.; Leak, R.K.; Hu, X.; Chen, J. Phagocytic microglia and macrophages in brain injury and repair. CNS Neurosci. Ther. 2022, 28, 1279–1293. [Google Scholar] [CrossRef]
- Hannestad, J.; Gallezot, J.-D.; Schafbauer, T.; Lim, K.; Kloczynski, T.; Morris, E.D.; Carson, R.E.; Ding, Y.-S.; Cosgrove, K.P. Endotoxin-induced systemic inflammation activates microglia: [11C]PBR28 positron emission tomography in nonhuman primates. NeuroImage 2012, 63, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflammation 2015, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Lemstra, A.W.; Hoozemans, J.J.; van Haastert, E.S.; Rozemuller, A.J.; Eikelenboom, P.; van Gool, W.A. Microglia activation in sepsis: A case-control study. J. Neuroinflammation 2007, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Michels, M.; Abatti, M.R.; Ávila, P.; Vieira, A.; Borges, H.; Junior, C.C.; Wendhausen, D.; Gasparotto, J.; Ribeiro, C.T.; Moreira, J.C.F.; et al. Characterization and modulation of microglial phenotypes in an animal model of severe sepsis. J. Cell. Mol. Med. 2020, 24, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Sandiego, C.M.; Gallezot, J.-D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.-F.; Matuskey, D.; Lee, J.-Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, D.; Engelen-Lee, J.Y.; Hoogland, I.C.M.; Aronica, E.M.A.; van Westerloo, D.J.; van de Beek, D.; van Gool, W.A. Systemic infection and microglia activation: A prospective postmortem study in sepsis patients. Immun. Ageing 2019, 16, 18. [Google Scholar] [CrossRef] [Green Version]
- Moraes, C.; Zaverucha-Do-Valle, C.; Fleurance, R.; Sharshar, T.; Bozza, F.; D’Avila, J. Neuroinflammation in Sepsis: Molecular Pathways of Microglia Activation. Pharmaceuticals 2021, 14, 416. [Google Scholar] [CrossRef] [PubMed]
- van Gool, W.A.; van de Beek, D.; Eikelenboom, P. Systemic infection and delirium: When cytokines and acetylcholine collide. Lancet 2010, 375, 773–775. [Google Scholar] [CrossRef]
- Michels, M.; Ávila, P.; Pescador, B.; Vieira, A.; Abatti, M.; Cucker, L.; Borges, H.; Goulart, A.I.; Junior, C.C.; Barichello, T.; et al. Microglial Cells Depletion Increases Inflammation and Modifies Microglial Phenotypes in an Animal Model of Severe Sepsis. Mol. Neurobiol. 2019, 56, 7296–7304. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Norenberg, M.D. Glutamine: A Trojan horse in ammonia neurotoxicity. Hepatology 2006, 44, 788–794. [Google Scholar] [CrossRef]
- Aldridge, D.R.; Tranah, E.J.; Shawcross, D.L. Pathogenesis of Hepatic Encephalopathy: Role of Ammonia and Systemic Inflammation. J. Clin. Exp. Hepatol. 2015, 5, S7–S20. [Google Scholar] [CrossRef] [Green Version]
- Jayakumar, A.R.; Rao, K.V.R.; Norenberg, M.D. Neuroinflammation in Hepatic Encephalopathy: Mechanistic Aspects. J. Clin. Exp. Hepatol. 2014, 5, S21–S28. [Google Scholar] [CrossRef] [Green Version]
- Thrane, V.R.; Thrane, A.; Chanag, J.; Alleluia, V.; Nagelhus, E.; Nedergaard, M. Real-time analysis of microglial activation and motility in hepatic and hyperammonemic encephalopathy. Neuroscience 2012, 220, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Zemtsova, I.; Görg, B.; Keitel, V.; Bidmon, H.-J.; Schrör, K.; Häussinger, D. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology 2011, 54, 204–215. [Google Scholar] [CrossRef]
- Jaeger, V.; DeMorrow, S.; McMillin, M. The Direct Contribution of Astrocytes and Microglia to the Pathogenesis of Hepatic Encephalopathy. J. Clin. Transl. Hepatol. 2019, 7, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Balzano, T.; Dadsetan, S.; Forteza, J.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Malaguarnera, M.; Gil-Perotin, S.; Cubas-Nuñez, L.; Casanova, B.; Castro-Quintas, A.; et al. Chronic hyperammonemia induces peripheral inflammation that leads to cognitive impairment in rats: Reversed by anti-TNF-α treatment. J. Hepatol. 2020, 73, 582–592. [Google Scholar] [CrossRef]
- Karababa, A.; Groos-Sahr, K.; Albrecht, U.; Keitel, V.; Shafigullina, A.; Görg, B.; Häussinger, D. Ammonia Attenuates LPS-Induced Upregulation of Pro-Inflammatory Cytokine mRNA in Co-Cultured Astrocytes and Microglia. Neurochem. Res. 2016, 42, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.V.R.; Brahmbhatt, M.; Norenberg, M.D. Microglia contribute to ammonia-induced astrocyte swelling in culture. Metab. Brain Dis. 2012, 28, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Shulyatnikova, T.; Shavrin, V. Mobilisation and redistribution of multivesicular bodies to the endfeet of reactive astrocytes in acute endogenous toxic encephalopathies. Brain Res. 2020, 1751, 147174. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod Microglia: A Morphological Definition. PLoS ONE 2014, 9, e97096. [Google Scholar] [CrossRef]
- Nissl, F. Über einige Beziehungen zwischen Nervenzellerkrankungen und gliösen Erscheinungen bei verschiedenen Psychosen. Arch. Psychiatr. 1899, 32, 656–676. [Google Scholar]
- Savage, J.C.; Carrier, M.; Tremblay, M.-È. Morphology of Microglia Across Contexts of Health and Disease. Methods Mol. Biol. 2019, 2034, 13–26. [Google Scholar] [CrossRef]
- Tremblay, M.-E.; Zettel, M.L.; Ison, J.R.; Allen, P.D.; Majewska, A.K. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 2012, 60, 541–558. [Google Scholar] [CrossRef] [Green Version]
- Augusto-Oliveira, M.; Arrifano, G.P.; Delage, C.I.; Tremblay, M.; Crespo-Lopez, M.E.; Verkhratsky, A. Plasticity of microglia. Biol. Rev. Cambridge Philos. Soc. 2021, 97, 217–250. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Van Eldik, L.J.; Schmitt, F.A.; Neltner, J.H.; Ighodaro, E.T.; Webster, S.J.; Patel, E.; Abner, E.L.; Kryscio, R.J.; Nelson, P.T. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol. Commun. 2015, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Ighodaro, E.T.; Hassoun, Y.; Aldeiri, D.; Neltner, J.H.; Patel, E.; Abner, E.L.; Nelson, P.T. Rod-shaped microglia morphology is associated with aging in 2 human autopsy series. Neurobiol. Aging 2017, 52, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Morrison, H.; Young, K.; Qureshi, M.; Rowe, R.; Lifshitz, J. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci. Rep. 2017, 7, 13211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, M.E.V.; Murray, H.C.; Ryan, B.; Faull, R.L.M.; Dragunow, M.; Curtis, M.A. Quantitative immunohistochemical analysis of myeloid cell marker expression in human cortex captures microglia heterogeneity with anatomical context. Sci. Rep. 2020, 10, 11693. [Google Scholar] [CrossRef] [PubMed]
- Dyne, E.; Cawood, M.; Suzelis, M.; Russell, R.; Kim, M. Ultrastructural analysis of the morphological phenotypes of microglia associated with neuroinflammatory cues. J. Comp. Neurol. 2022, 530, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, H.; Savage, J.C.; Bisht, K.; Parent, M.; Vallières, L.; Rivest, S.; Tremblay, M. Ultrastructural evidence of microglial heterogeneity in Alzheimer’s disease amyloid pathology. J. Neuroinflammation 2019, 16, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, K.; Svensson, L.A.; Mohlin, C. Morphological analyzes of microglia heterogeneity and dynamics during photoreceptor degeneration in vitro: Presumptive dark microglia in porcine retina. Exp. Eye Res. 2020, 200, 108217. [Google Scholar] [CrossRef]
- Krukowski, K.; Nolan, A.; Becker, M.; Picard, K.; Vernoux, N.; Frias, E.S.; Feng, X.; Tremblay, M.-E.; Rosi, S. Novel microglia-mediated mechanisms underlying synaptic loss and cognitive impairment after traumatic brain injury. Brain Behav. Immun. 2021, 98, 122–135. [Google Scholar] [CrossRef]
- Savage, J.C.; St-Pierre, M.-K.; Hui, C.W.; Tremblay, M.-E. Microglial Ultrastructure in the Hippocampus of a Lipopolysaccharide-Induced Sickness Mouse Model. Front. Neurosci. 2019, 13, 1340. [Google Scholar] [CrossRef] [Green Version]
- Zrzavy, T.; Höftberger, R.; Berger, T.; Rauschka, H.; Butovsky, O.; Weiner, H.; Lassmann, H. Pro-inflammatory activation of microglia in the brain of patients with sepsis. Neuropathol. Appl. Neurobiol. 2018, 45, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lin, F.; Ren, M.; Liu, X.; Xie, W.; Zhang, A.; Qian, M.; Mo, Y.; Wang, J.; Lv, Y. The PICK1/TLR4 complex on microglia is involved in the regulation of LPS-induced sepsis-associated encephalopathy. Int. Immunopharmacol. 2021, 100, 108116. [Google Scholar] [CrossRef]
- Shulyatnikova, T.V.; Shavrin, V.O. Regional-specific activation of phagocytosis in the rat brain in the conditions of sepsis-associated encephalopathy. Zaporozhye Med. J. 2021, 23, 111–119. [Google Scholar] [CrossRef]
- Kaur, C.; Ling, E.A.; Wong, W.C. Transformation of amoeboid microglial cells into microglia in the corpus callosum of the postnatal rat brain. An electron microscopical study. Arch. Histol. Jpn. 1985, 48, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, C.; Rathnasamy, G.; Ling, E.-A. Biology of Microglia in the Developing Brain. J. Neuropathol. Exp. Neurol. 2017, 76, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Ling, E.; Wong, W. The origin and nature of ramified and amoeboid microglia: A historical review and current concepts. Glia 1993, 7, 9–18. [Google Scholar] [CrossRef]
- Stence, N.; Waite, M.; Dailey, M.E. Dynamics of microglial activation: A confocal time-lapse analysis in hippocampal slices. Glia 2001, 33, 256–266. [Google Scholar] [CrossRef]
- Berglund, R.; Guerreiro-Cacais, A.O.; Adzemovic, M.Z.; Zeitelhofer, M.; Lund, H.; Ewing, E.; Ruhrmann, S.; Nutma, E.; Parsa, R.; Thessen-Hedreul, M.; et al. Microglial autophagy–associated phagocytosis is essential for recovery from neuroinflammation. Sci. Immunol. 2020, 5, eabb5077. [Google Scholar] [CrossRef]
- Jia, J.; Yang, L.; Chen, Y.; Zheng, L.; Chen, Y.; Xu, Y.; Zhang, M. The Role of Microglial Phagocytosis in Ischemic Stroke. Front. Immunol. 2022, 12, 5704. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.; Tang, Y. Phagocytosis of Microglia in the Central Nervous System Diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Lu, J.; Sivakumar, V.; Ling, E.A.; Kaur, C. Amoeboid Microglia in the Periventricular White Matter Induce Oligodendrocyte Damage through Expression of Proinflammatory Cytokines via MAP Kinase Signaling Pathway in Hypoxic Neonatal Rats. Brain Pathol. 2008, 18, 387–400. [Google Scholar] [CrossRef]
- Henry, C.J.; Huang, Y.; Wynne, A.M.; Godbout, J.P. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain, Behav. Immun. 2009, 23, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Hoogland, I.C.M.; Westhoff, D.; Engelen-Lee, J.-Y.; Melief, J.; Serón, M.V.; Houben-Weerts, J.H.M.P.; Huitinga, I.; Van Westerloo, D.J.; Van Der Poll, T.; Van Gool, W.A.; et al. Microglial Activation After Systemic Stimulation with Lipopolysaccharide and Escherichia coli. Front. Cell. Neurosci. 2018, 12, 110. [Google Scholar] [CrossRef] [Green Version]
- Shavrin, V.A.; Shulyatnikova, T.; Polkovnikov, Y.F. Morphogenesis of the “ischaemic-homogenizing” neuronal changes and their significance in the pathohistological diagnostics of ischaemic diseases of the brain. Pathologia 2008, 5, 73–78. [Google Scholar]
- Kafa, I.M.; Uysal, M.; Bakirci, S.; Kurt, M.A. Sepsis induces apoptotic cell death in different regions of the brain in a rat model of sepsis. Acta Neurobiol. Exp. 2010, 70, 246–260. [Google Scholar]
- Sharshar, T.; Gray, F.; de la Grandmaison, G.L.; Hopklnson, N.S.; Ross, E.; Dorandeu, A.; Orlikowski, D.; Raphael, J.-C.; Gajdos, P.; Annane, D. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet 2003, 362, 1799–1805. [Google Scholar] [CrossRef]
- Sharshar, T.; Annane, D.; De La Grandmaison, G.L.; Brouland, J.P.; Hopkinson, N.; Gray, F. The Neuropathology of Septic Shock. Brain Pathol. 2004, 14, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Cserép, C.; Pósfai, B.; Dénes, Á. Shaping Neuronal Fate: Functional Heterogeneity of Direct Microglia-Neuron Interactions. Neuron 2020, 109, 222–240. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2012, 61, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Nebeling, F.C.; Poll, S.; Schmid, L.C.; Mittag, M.; Steffen, J.; Keppler, K.; Fuhrmann, M. Microglia motility depends on neuronal activity and promotes structural plasticity in the hippocampus. bioRxiv 2019. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Bystrom, L.L.; Ying, Y.; Liu, Y.U.; Worrell, G.; Wu, L.-J. Microglial calcium signaling is attuned to neuronal activity in awake mice. eLife 2020, 9, e56502. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.M.; Fekete, R.; Horvath, G.; Koncsos, G.; Kriston, C.; Sebestyen, A.; Giricz, Z.; Kornyei, Z.; Madarasz, E.; Tretter, L. Versatility of microglial bioenergetic machinery under starving conditions. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2018, 1859, 201–214. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madore, C.; Joffre, C.; Delpech, J.; De Smedt-Peyrusse, V.; Aubert, A.; Coste, L.; Layé, S.; Nadjar, A. Early morphofunctional plasticity of microglia in response to acute lipopolysaccharide. Brain Behav. Immun. 2013, 34, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, A.; Salm, A.K. Increased Morphological Diversity of Microglia in the Activated Hypothalamic Supraoptic Nucleus. J. Neurosci. 2003, 23, 7759–7766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parakalan, R.; Jiang, B.; Nimmi, B.; Janani, M.; Jayapal, M.; Lu, J.; Tay, S.S.; Ling, E.-A.; Dheen, S.T. Transcriptome analysis of amoeboid and ramified microglia isolated from the corpus callosum of rat brain. BMC Neurosci. 2012, 13, 64. [Google Scholar] [CrossRef]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.-F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Oh, E.; Yun, M.; Lee, S.-B.; Chae, G.T. Palmitate induces cisternal ER expansion via the activation of XBP-1/CCTα-mediated phospholipid accumulation in RAW 264.7 cells. Lipids Health Dis. 2015, 14, 73. [Google Scholar] [CrossRef] [Green Version]
- Kacimi, R.; Giffard, R.G.; Yenari, M.A. Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB, JAK-STAT and JNK stress kinase pathways. J. Inflamm. 2011, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, L.P.; Hora, A.S.; De Siqueira, A.; Salvagni, F.A.; Brandão, P.E.; Maiorka, P.C. Glial response in the central nervous system of cats with feline infectious peritonitis. J. Feline Med. Surg. 2016, 18, 1023–1030. [Google Scholar] [CrossRef]
- Tremblay, M.-E.; Zhang, I.; Bisht, K.; Savage, J.C.; Lecours, C.; Parent, M.; Titorenko, V.; Maysinger, D. Remodeling of lipid bodies by docosahexaenoic acid in activated microglial cells. J. Neuroinflammation 2016, 13, 116. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Nosi, D.; Lana, D.; Giovannini, M.; Delfino, G.; Zecchi-Orlandini, S. Neuroinflammation: Integrated Nervous Tissue Response through Intercellular Interactions at the “Whole System” Scale. Cells 2021, 10, 1195. [Google Scholar] [CrossRef] [PubMed]
- Shulyatnikova, T.V. Immunohistochemical analysis of microglial changes in the experimental acute hepatic encephalopathy. Pathologia 2021, 18, 33–38. [Google Scholar] [CrossRef]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Buckwalter, M.S. Astrocytes: Integrative Regulators of Neuroinflammation in Stroke and Other Neurological Diseases. Neurotherapeutics 2016, 13, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Font-Nieves, M.; Sans-Fons, M.G.; Gorina, R.; Bonfill-Teixidor, E.; SalasPerdomo, A.; Marquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Induction of COX-2 Enzyme and Down-regulation of COX-1 Expression by Lipopolysaccharide (LPS) Control Prostaglandin E2 Production in Astrocytes. J. Biol. Chem. 2012, 287, 6454–6468. [Google Scholar] [CrossRef] [Green Version]
- Coltart, I.; Tranah, T.H.; Shawcross, D.L. Inflammation and hepatic encephalopathy. Arch. Biochem. Biophys. 2013, 536, 189–196. [Google Scholar] [CrossRef]
- Odeh, M. Endotoxin and Tumor Necrosis Factor-α in the Pathogenesis of Hepatic Encephalopathy. J. Clin. Gastroenterol. 1994, 19, 146–153. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Wierzba-Bobrowicz, T.; Gwiazda, E.; Kosno-Kruszewska, E.; Lewandowska, E.; Lechowicz, W.; Bertrand, E.; Szpak, G.M.; Schmidt-Sidor, B. Morphological analysis of active microglia—Rod and ramified microglia in human brains affected by some neurological diseases (SSPE, Alzheimer’s disease and Wilson’s disease). Folia Neuropathol. 2002, 40, 125–131. [Google Scholar]
- Wirenfeldt, M.; Clare, R.; Tung, S.; Bottini, A.; Mathern, G.W.; Vinters, H.V. Increased activation of Iba1+ microglia in pediatric epilepsy patients with Rasmussen’s encephalitis compared with cortical dysplasia and tuberous sclerosis complex. Neurobiol. Dis. 2009, 34, 432–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondo, E.; Becker, S.C.; Kautzman, A.G.; Schifferer, M.; Baer, C.E.; Chen, J.; Huang, E.J.; Simons, M.; Schafer, D.P. A Developmental Analysis of Juxtavascular Microglia Dynamics and Interactions with the Vasculature. J. Neurosci. 2020, 40, 6503–6521. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, R.; Stence, N.; Carr, J.; Fuller, L.; Waite, M.; Dailey, M.E. Juxtavascular microglia migrate along brain microvessels following activation during early postnatal development. Glia 2002, 37, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; Demarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model–Part II: Microglia and Mitochondria. Neuroglia 2018, 1, 311–326. [Google Scholar] [CrossRef]
- Sun, Y.; Koyama, Y.; Shimada, S. Inflammation from Peripheral Organs to the Brain: How Does Systemic Inflammation Cause Neuroinflammation? Front. Aging Neurosci. 2022, 14, 903455. [Google Scholar] [CrossRef]
- Hayden, M.R. Hypothesis: Neuroglia Activation Due to Increased Peripheral and CNS Proinflammatory Cytokines/Chemokines with Neuroinflammation May Result in Long COVID. Neuroglia 2021, 2, 7–35. [Google Scholar] [CrossRef]
- Hayden, M.R. Overview of Neuroglia Activation, Chronic Neuroinflammation, Remodeling, and Impaired Cognition Due to Perivascular Adipose Tissue-Derived Extracellular Vesicle Exosomes in Obesity and Diabetes. Neuroglia 2022, 3, 112–138. [Google Scholar] [CrossRef]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef]
- McGill, M.R.; Williams, C.D.; Xie, Y.; Ramachandran, A.; Jaeschke, H. Acetaminophen-induced liver injury in rats and mice: Comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol. Appl. Pharmacol. 2012, 264, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Mossanen, J.C.; Tacke, F. Acetaminophen-induced acute liver injury in mice. Lab. Anim. 2015, 49, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.R.; Marsden, P. A rapid methylene blue-basic fuchsin stain for semi-thin sections of peripheral nerve and other tissues. J. Microsc. 1969, 89, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.A.; Perez, Z.D.; Foresti, M.L.; Arisi, G.M.; Ribak, C.E. Morphological and ultrastructural features of Iba1-immunolabeled microglial cells in the hippocampal dentate gyrus. Brain Res. 2009, 1266, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinsen, H. Lipofuscin in the cerebellar cortex of albino rats: An electron microscopic study. Anat. Embryol. 1979, 155, 333–345. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shulyatnikova, T.; Tumanskyi, V.; Hayden, M.R. Reactive Microgliosis in Sepsis-Associated and Acute Hepatic Encephalopathies: An Ultrastructural Study. Int. J. Mol. Sci. 2022, 23, 14455. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214455
Shulyatnikova T, Tumanskyi V, Hayden MR. Reactive Microgliosis in Sepsis-Associated and Acute Hepatic Encephalopathies: An Ultrastructural Study. International Journal of Molecular Sciences. 2022; 23(22):14455. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214455
Chicago/Turabian StyleShulyatnikova, Tatyana, Valerii Tumanskyi, and Melvin R. Hayden. 2022. "Reactive Microgliosis in Sepsis-Associated and Acute Hepatic Encephalopathies: An Ultrastructural Study" International Journal of Molecular Sciences 23, no. 22: 14455. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214455