
Sphingosine Kinase 1 Deficiency in Smooth Muscle Cells Protects against Hypoxia-Mediated Pulmonary Hypertension via YAP1 Signaling
 , , , , , and
, , , , , and 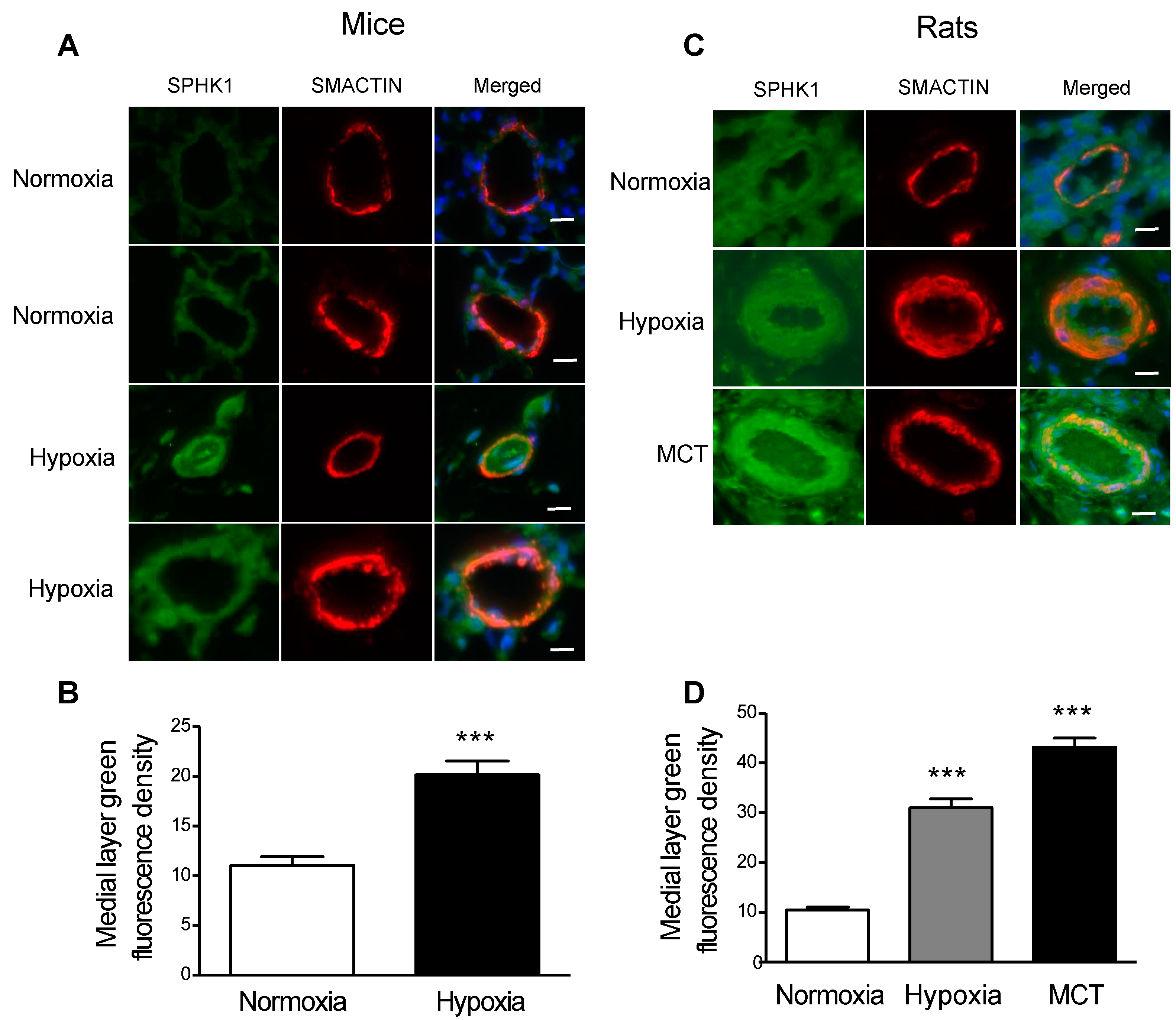{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. SPHK1 Is Upregulated in the PASMCs of Rodent Models of PH
2.2. Smooth Muscle Cell-Specific Sphk1 Knockout Mice Are Protected from Hypoxia-Induced PH
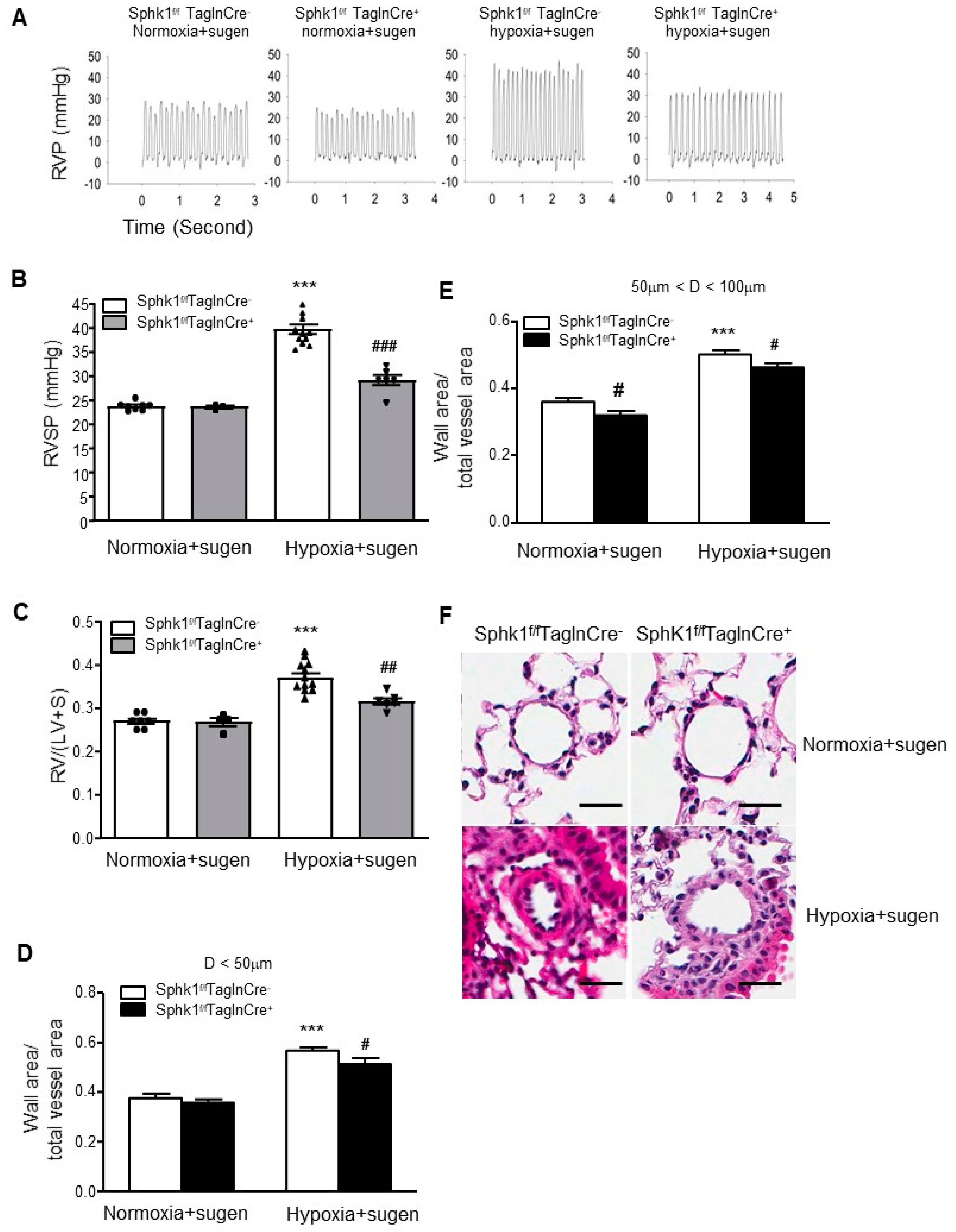
2.3. Smooth Muscle Cell-Specific Sphk1 Knockout Mice Are Protected from Hypoxia Plus Sugen-Mediated Pulmonary Hypertension
2.4. PASMCs Isolated from Sphk1-/- Mice Are Less Proliferative and Less Viable
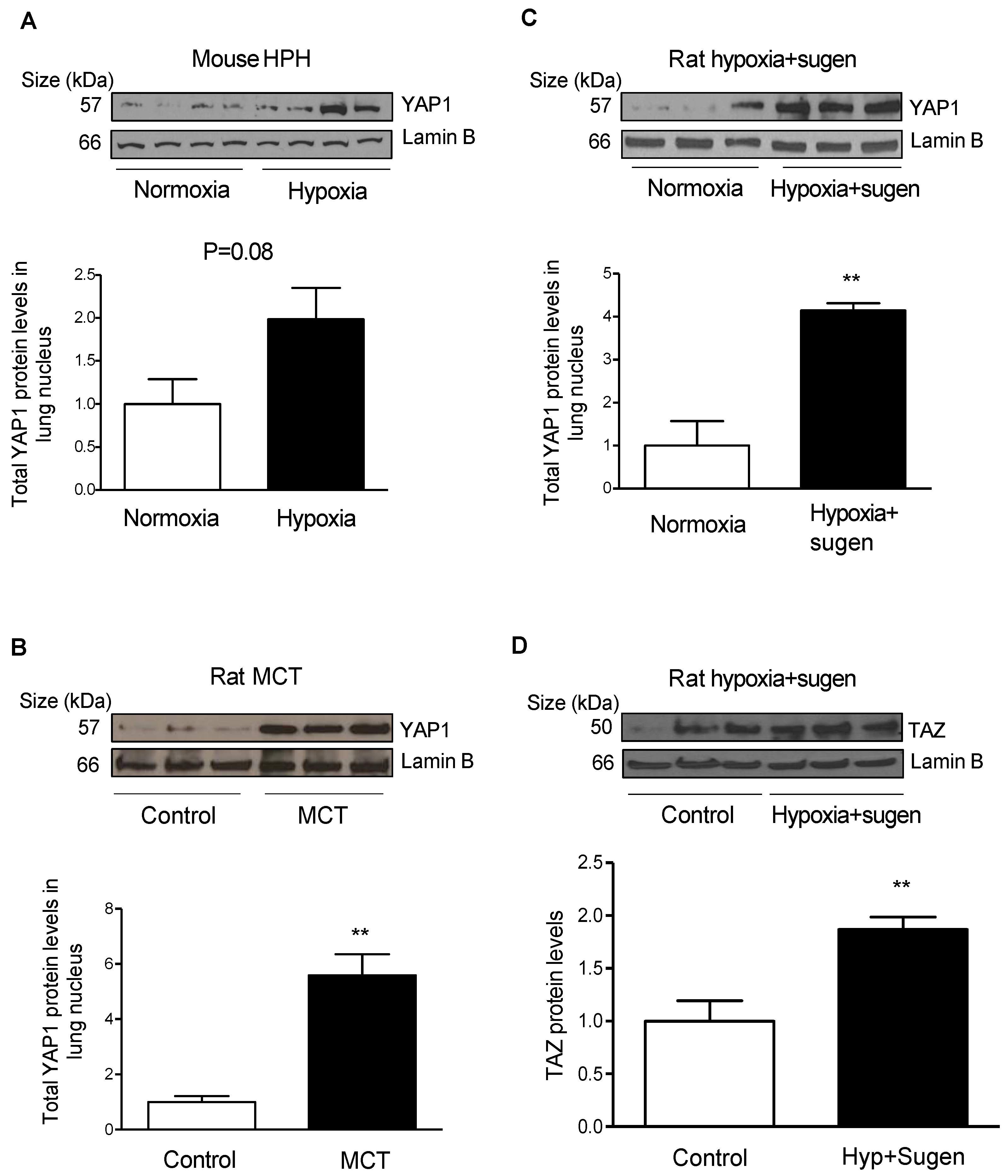
2.5. Hypoxia Induces YAP1 and TAZ Expression in the Lungs of Rodent Models of PH
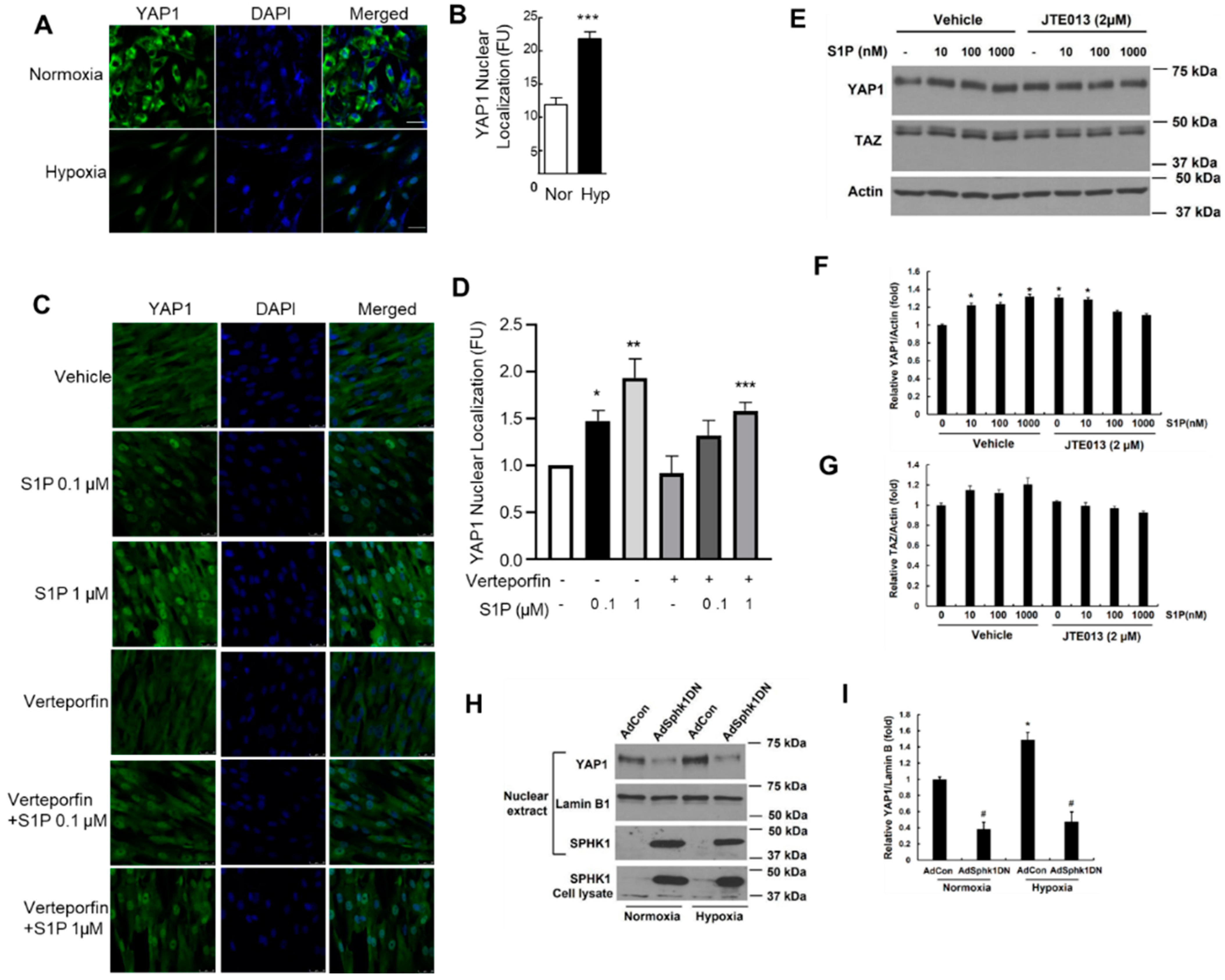
2.6. Hypoxia and S1P Stimulate YAP1 Nuclear Translocation in PASMCs
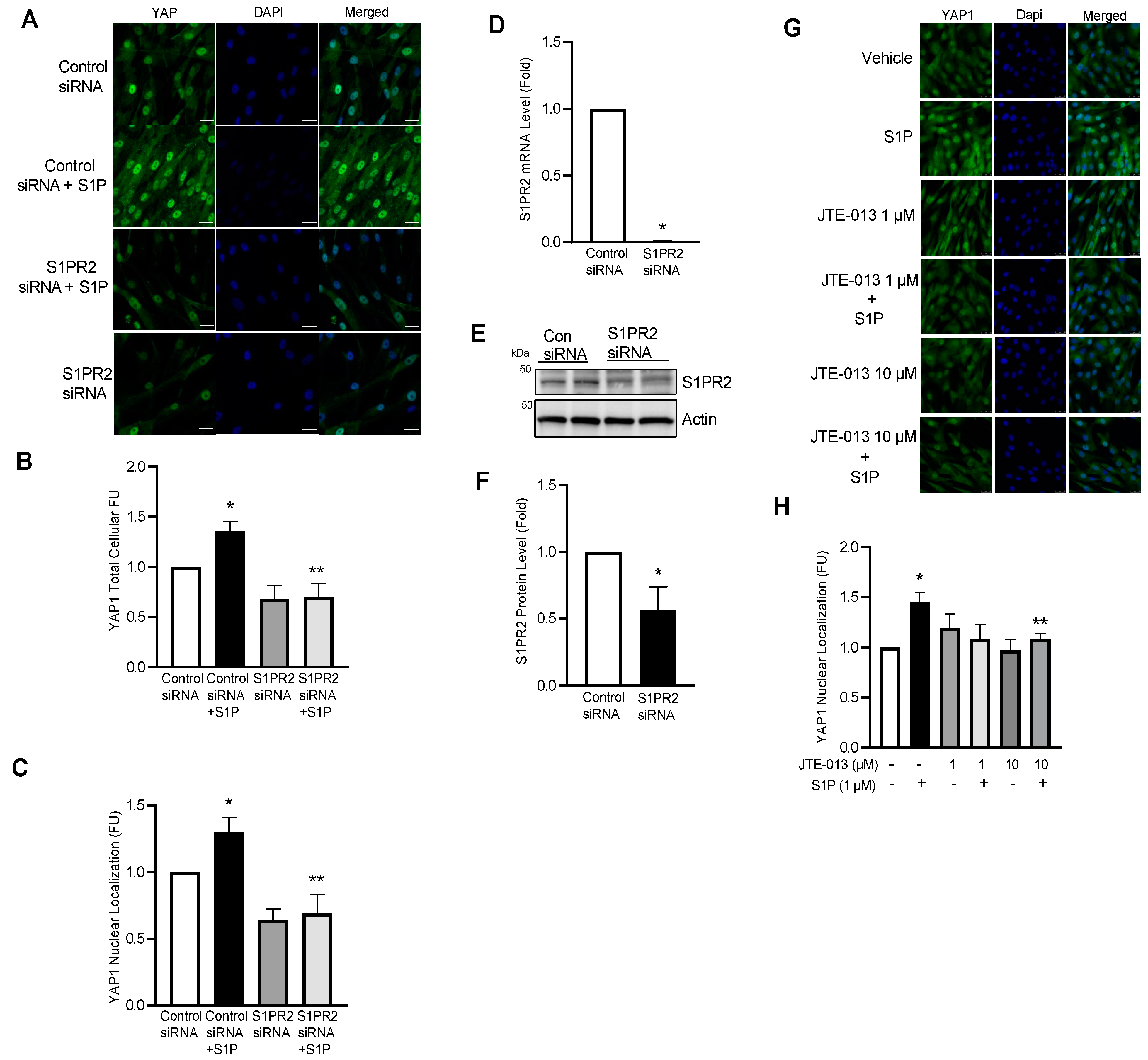
2.7. S1P Promotes YAP1 Nuclear Translocation via Ligation to S1PR2
2.8. YAP1 Regulates S1P-Mediated Proliferation in hPASMCs
2.9. YAP1 Inhibition with Verteporfin Prevents Hypoxia-Mediated PH and Pulmonary Vascular Remodeling in Mice
2.10. YAP1 Inhibition with Verteporfin Attenuates Hypoxia/Sugen-Mediated PH and Pulmonary Vascular Remodeling in Rats
3. Discussion
4. Materials and Methods
4.1. Reagents, Drugs, and Antibodies
4.2. Mouse PASMC Isolation
4.3. Cell Proliferation, Viability, Apoptosis, and Lactate Dehydrogenase Assays
4.4. Lung Tissue Immunofluorescence Staining
4.5. Adenoviral Constructs of Wild-Type and Dominant-Negative SPHK1 Plasmids
4.6. Transfection of Plasmid DNA and Small Interfering RNA in hPASMCs
4.7. Animal Models of Pulmonary Hypertension and Hemodynamic Measurements
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Sysol, J.R.; Singla, S.; Zhao, S.; Yamamura, A.; Valdez-Jasso, D.; Abbasi, T.; Shioura, K.M.; Sahni, S.; Reddy, V.; et al. Nicotinamide Phosphoribosyltransferase Promotes Pulmonary Vascular Remodeling and Is a Therapeutic Target in Pulmonary Arterial Hypertension. Circulation 2017, 135, 1532–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, R.; Abassi, T.; Machado, R.F.; Chen, J. Treatment of Idiopathic Pulmonary Hypertension: Current and Clinical Trial Modalities. J. Hypertens. Manag. 2016, 2, 5. [Google Scholar] [CrossRef]
- Yuan, J.X.; Rubin, L.J. Pathogenesis of pulmonary arterial hypertension: The need for multiple hits. Circulation 2005, 111, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.S.; Berdyshev, E.; Mathew, B.; Fu, P.; Gorshkova, I.A.; He, D.; Ma, W.; Noth, I.; Ma, S.F.; Pendyala, S.; et al. Targeting sphingosine kinase 1 attenuates bleomycin-induced pulmonary fibrosis. FASEB J. 2013, 27, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef] [Green Version]
- Olivera, A.; Kohama, T.; Edsall, L.; Nava, V.; Cuvillier, O.; Poulton, S.; Spiegel, S. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J. Cell Biol. 1999, 147, 545–558. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.; Yang, J.; DeRan, M.; Wu, C.; Su, A.I.; Bonamy, G.M.; Liu, J.; Peters, E.C.; Wu, X. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol. 2012, 19, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhai, C.; Pan, Y.; Zhu, Y.; Shi, W.; Wang, J.; Yan, X.; Su, X.; Song, Y.; Gao, L.; et al. Sphingosine-1-phosphate induces airway smooth muscle cell proliferation, migration, and contraction by modulating Hippo signaling effector YAP. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L609–L621. [Google Scholar] [CrossRef] [Green Version]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieffenbach, P.B.; Haeger, C.M.; Coronata, A.M.F.; Choi, K.M.; Varelas, X.; Tschumperlin, D.J.; Fredenburgh, L.E. Arterial stiffness induces remodeling phenotypes in pulmonary artery smooth muscle cells via YAP/TAZ-mediated repression of cyclooxygenase-2. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L628–L647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, T.V.; Goncharov, D.A.; Pena, A.; Kelly, N.; Vanderpool, R.; Baust, J.; Kobir, A.; Shufesky, W.; Mora, A.L.; Morelli, A.E.; et al. HIPPO-Integrin-linked Kinase Cross-Talk Controls Self-Sustaining Proliferation and Survival in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell. 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Zhao, Y.; Kalari, S.K.; Usatyuk, P.V.; Gorshkova, I.; He, D.; Watkins, T.; Brindley, D.N.; Sun, C.; Bittman, R.; Garcia, J.G.; et al. Intracellular generation of sphingosine 1-phosphate in human lung endothelial cells: Role of lipid phosphate phosphatase-1 and sphingosine kinase 1. J. Biol. Chem. 2007, 282, 14165–14177. [Google Scholar] [CrossRef] [Green Version]
- Sysol, J.R.; Chen, J.; Singla, S.; Zhao, S.; Comhair, S.; Natarajan, V.; Machado, R.F. Micro-RNA-1 is decreased by hypoxia and contributes to the development of pulmonary vascular remodeling via regulation of sphingosine kinase 1. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L461–L472. [Google Scholar] [CrossRef]
- Sysol, J.R.; Natarajan, V.; Machado, R.F. PDGF induces SphK1 expression via Egr-1 to promote pulmonary artery smooth muscle cell proliferation. Am. J. Physiol. Cell Physiol. 2016, 310, C983–C992. [Google Scholar] [CrossRef] [Green Version]
- Chai, S.; Wang, W.; Liu, J.; Guo, H.; Zhang, Z.; Wang, C.; Wang, J. Leptin knockout attenuates hypoxia-induced pulmonary arterial hypertension by inhibiting proliferation of pulmonary arterial smooth muscle cells. Transl. Res. 2015, 166, 772–782. [Google Scholar] [CrossRef]
- Gairhe, S.; Joshi, S.R.; Bastola, M.M.; McLendon, J.M.; Oka, M.; Fagan, K.A.; McMurtry, I.F. Sphingosine-1-phosphate is involved in the occlusive arteriopathy of pulmonary arterial hypertension. Pulm Circ. 2016, 6, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.C.; Wang, E.Y.; Yi, Y.; Thakur, A.; Tsai, S.H.; Hoodless, P.A. S1P Stimulates Proliferation by Upregulating CTGF Expression through S1PR2-Mediated YAP Activation. Mol. Cancer Res. 2018, 16, 1543–1555. [Google Scholar] [CrossRef]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Wang, Q.; Wang, J.; Yan, X.; Feng, W.; Zhang, Q.; Zhai, C.; Chai, L.; Li, S.; Xie, X.; et al. Activation of yes-associated protein mediates sphingosine-1-phosphate-induced proliferation and migration of pulmonary artery smooth muscle cells and its potential mechanisms. J. Cell Physiol. 2021, 236, 4694–4708. [Google Scholar] [CrossRef]
- Wang, J.; Yan, X.; Feng, W.; Wang, Q.; Shi, W.; Chai, L.; Zhang, Q.; Chen, Y.; Liu, J.; Qu, Z.; et al. S1P induces proliferation of pulmonary artery smooth muscle cells by promoting YAP-induced Notch3 expression and activation. J. Biol. Chem. 2021, 296, 100599. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shi, W.; Zhang, Q.; Feng, W.; Wang, J.; Zhai, C.; Yan, X.; Li, M. Inhibition of Siah2 ubiquitin ligase ameliorates monocrotaline-induced pulmonary arterial remodeling through inactivation of YAP. Life Sci. 2020, 242, 117159. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.; Mamary, A.J.; Verhoeven, A.J.; Marshall, B.E. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am. J. Respir. Cell Mol. Biol. 1996, 15, 633–644. [Google Scholar] [CrossRef]
- Ciuclan, L.; Bonneau, O.; Hussey, M.; Duggan, N.; Holmes, A.M.; Good, R.; Stringer, R.; Jones, P.; Morrell, N.W.; Jarai, G.; et al. A novel murine model of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2011, 184, 1171–1182. [Google Scholar] [CrossRef]
- Ma, W.; Han, W.; Greer, P.A.; Tuder, R.M.; Toque, H.A.; Wang, K.K.; Caldwell, R.W.; Su, Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. J. Clin. Investig. 2011, 121, 4548–4566. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Lockett, A.; Zhao, S.; Huang, L.S.; Wang, Y.; Wu, W.; Tang, M.; Haider, S.; Velez Rendon, D.; Khan, R.; et al. Sphingosine Kinase 1 Deficiency in Smooth Muscle Cells Protects against Hypoxia-Mediated Pulmonary Hypertension via YAP1 Signaling. Int. J. Mol. Sci. 2022, 23, 14516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232314516
Chen J, Lockett A, Zhao S, Huang LS, Wang Y, Wu W, Tang M, Haider S, Velez Rendon D, Khan R, et al. Sphingosine Kinase 1 Deficiency in Smooth Muscle Cells Protects against Hypoxia-Mediated Pulmonary Hypertension via YAP1 Signaling. International Journal of Molecular Sciences. 2022; 23(23):14516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232314516
Chicago/Turabian StyleChen, Jiwang, Angelia Lockett, Shuangping Zhao, Long Shuang Huang, Yifan Wang, Weiwen Wu, Ming Tang, Shahzaib Haider, Daniela Velez Rendon, Raheel Khan, and et al. 2022. "Sphingosine Kinase 1 Deficiency in Smooth Muscle Cells Protects against Hypoxia-Mediated Pulmonary Hypertension via YAP1 Signaling" International Journal of Molecular Sciences 23, no. 23: 14516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232314516