
Cancer Stem Cell Formation Induced and Regulated by Extracellular ATP and Stanniocalcin-1 in Human Lung Cancer Cells and Tumors

 , and
, and
Abstract
:1. Introduction
2. Results
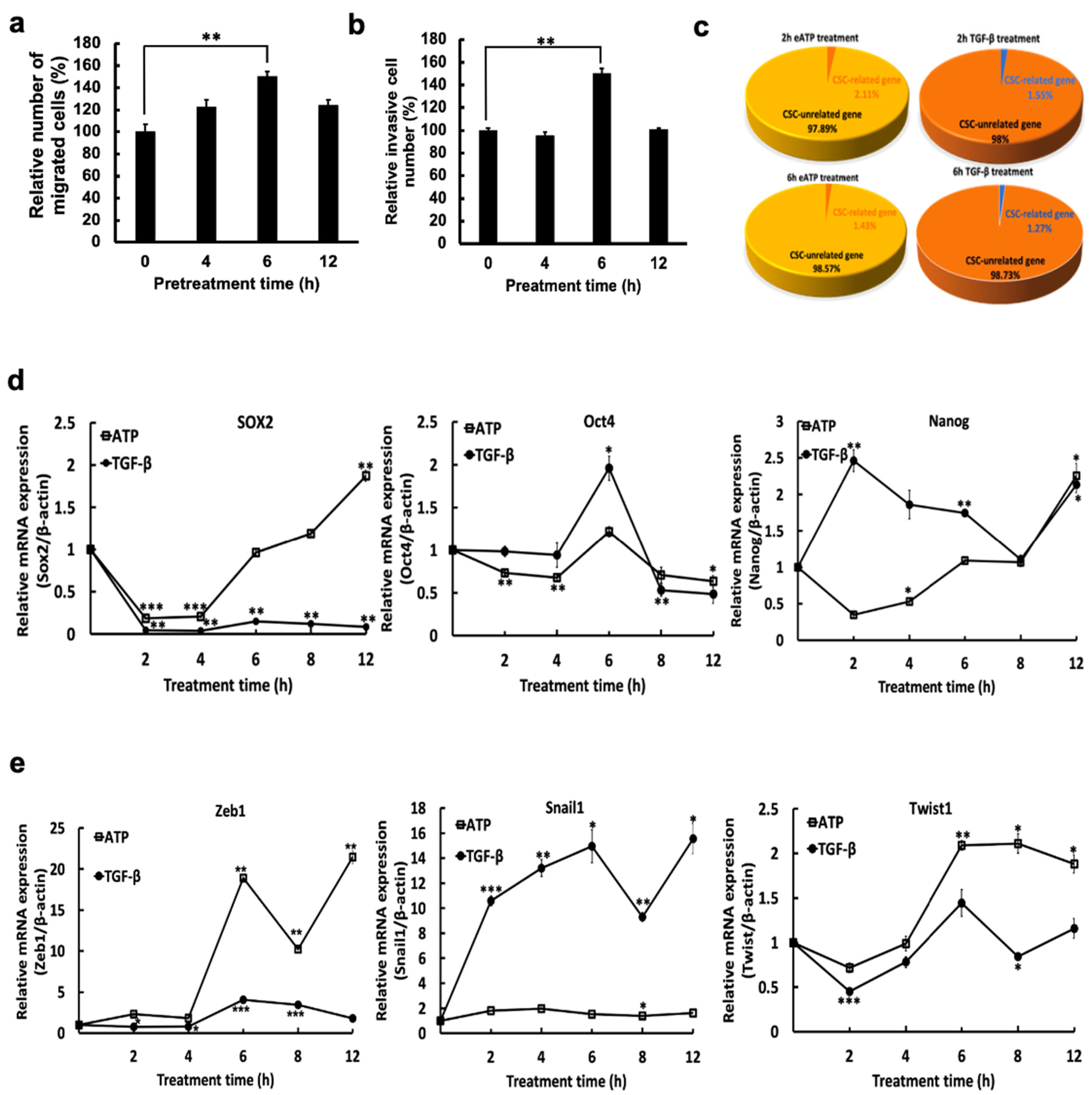
2.1. eATP Increased Rates of Cell Migration and Invasion and Altered Expression of Genes Involved in CSCs
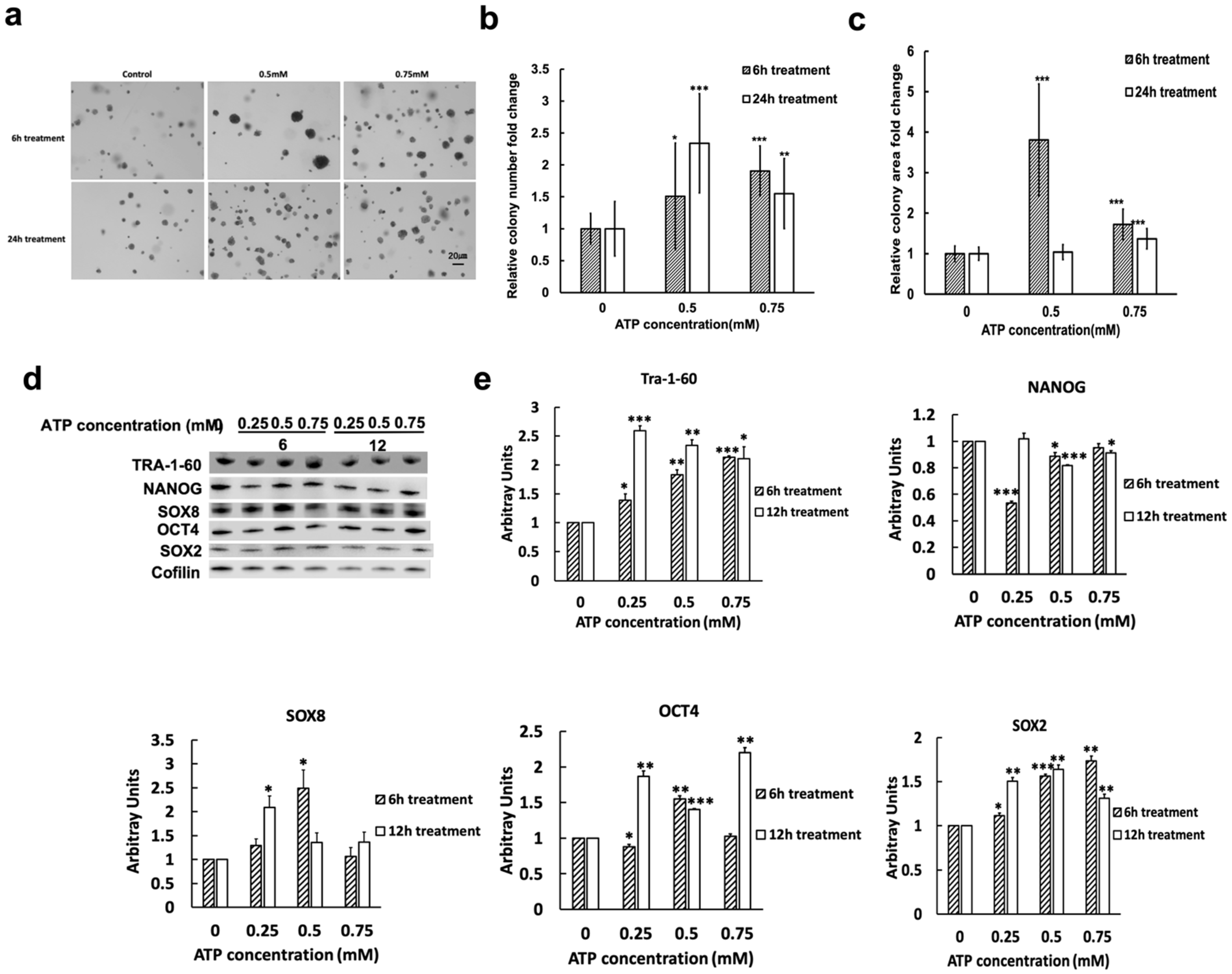
2.2. eATP Induced Time- and Dose-Dependent Changes in Colony Formation and Levels of Proteins Involved in CSC Formation
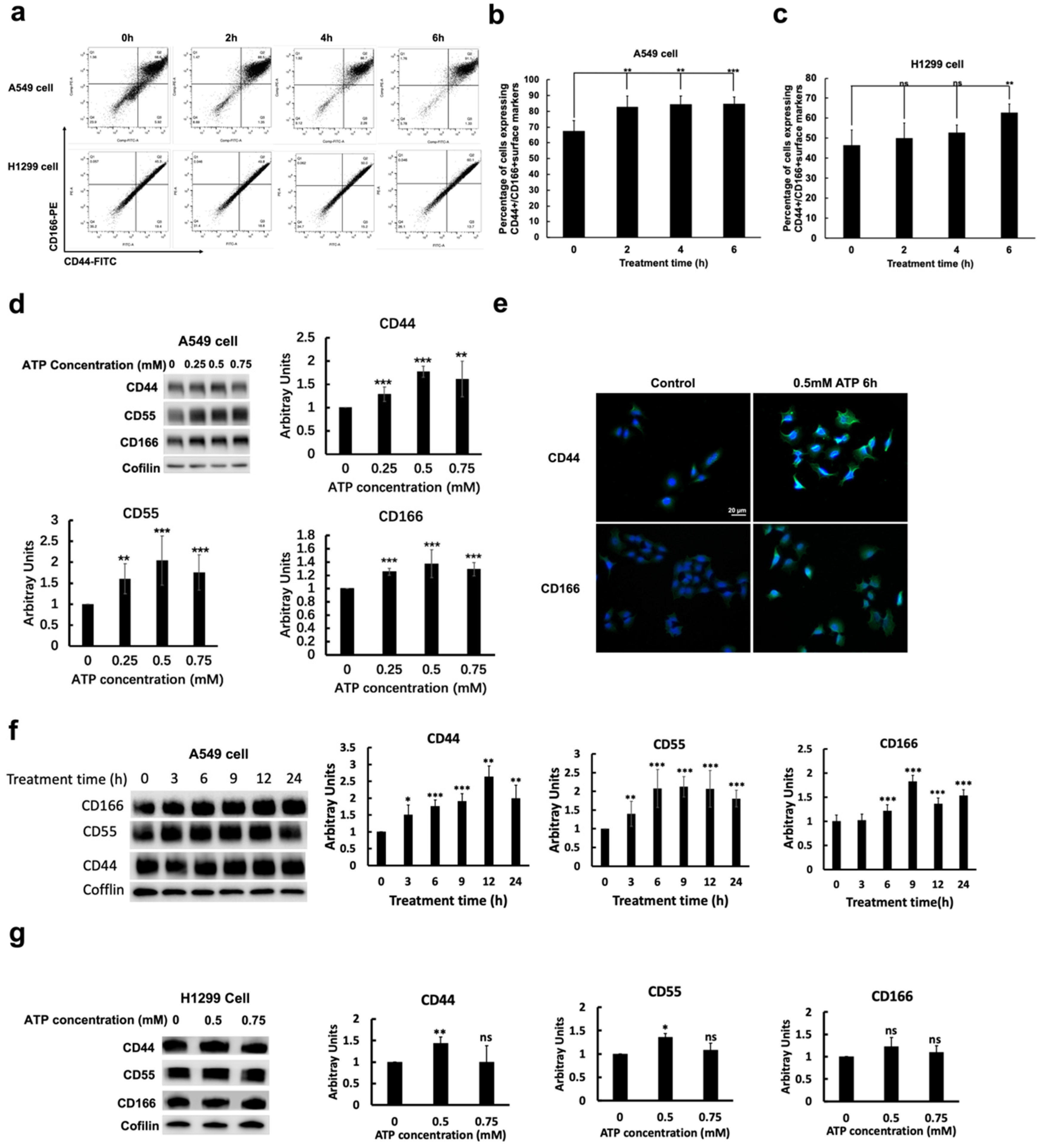
2.3. eATP Increased CSC Surface Markers and CSC Subpopulations in NSCLC Cells
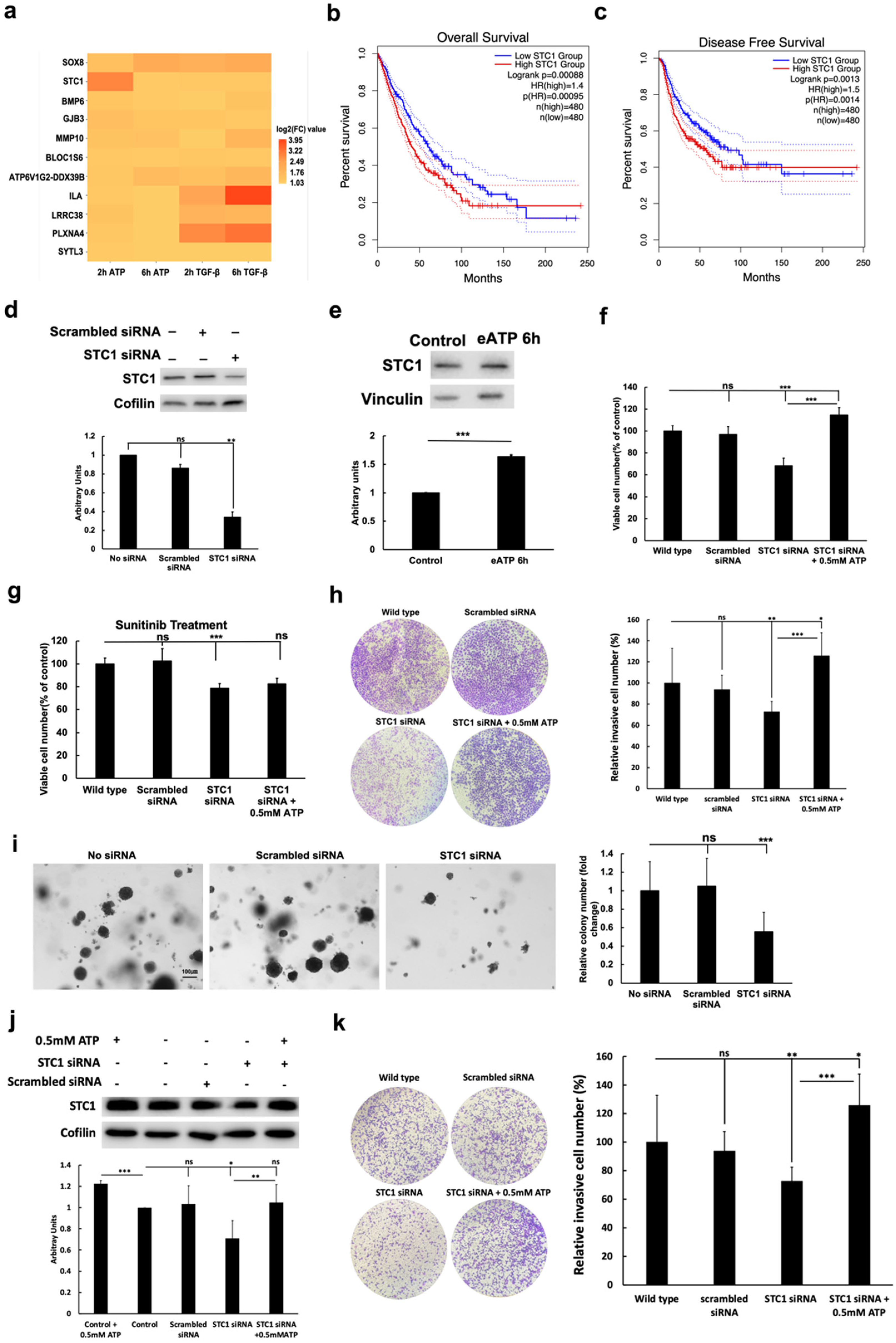
2.4. eATP Induced Genes Involved in EMT and CSC, and KD of STC1 Reduced CSC Phenotypic Changes
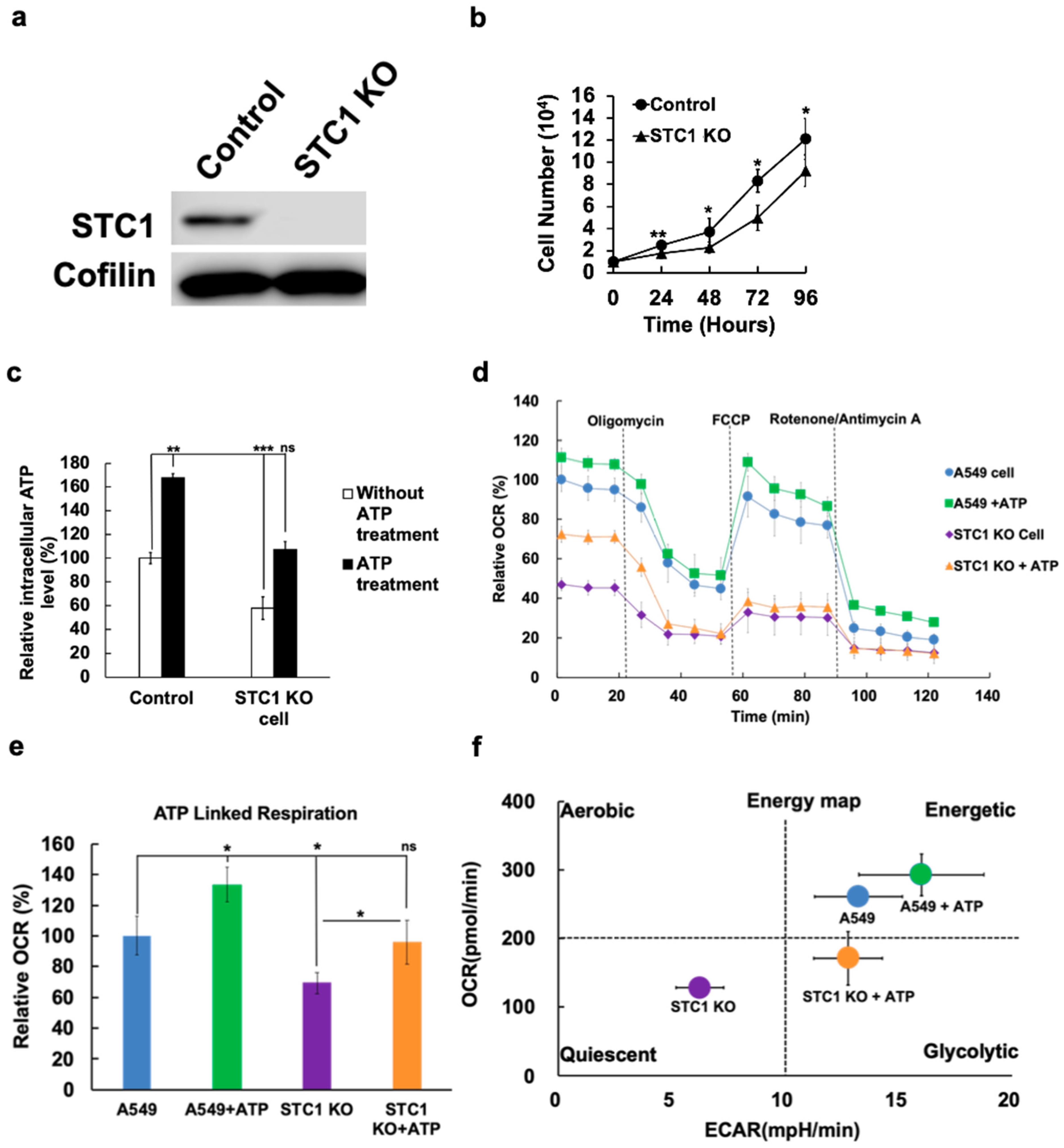
2.5. Knockout of STC1 Lowered iATP Levels, Oxygen Consumption Rate, and Mitochondrial ATP Synthesis
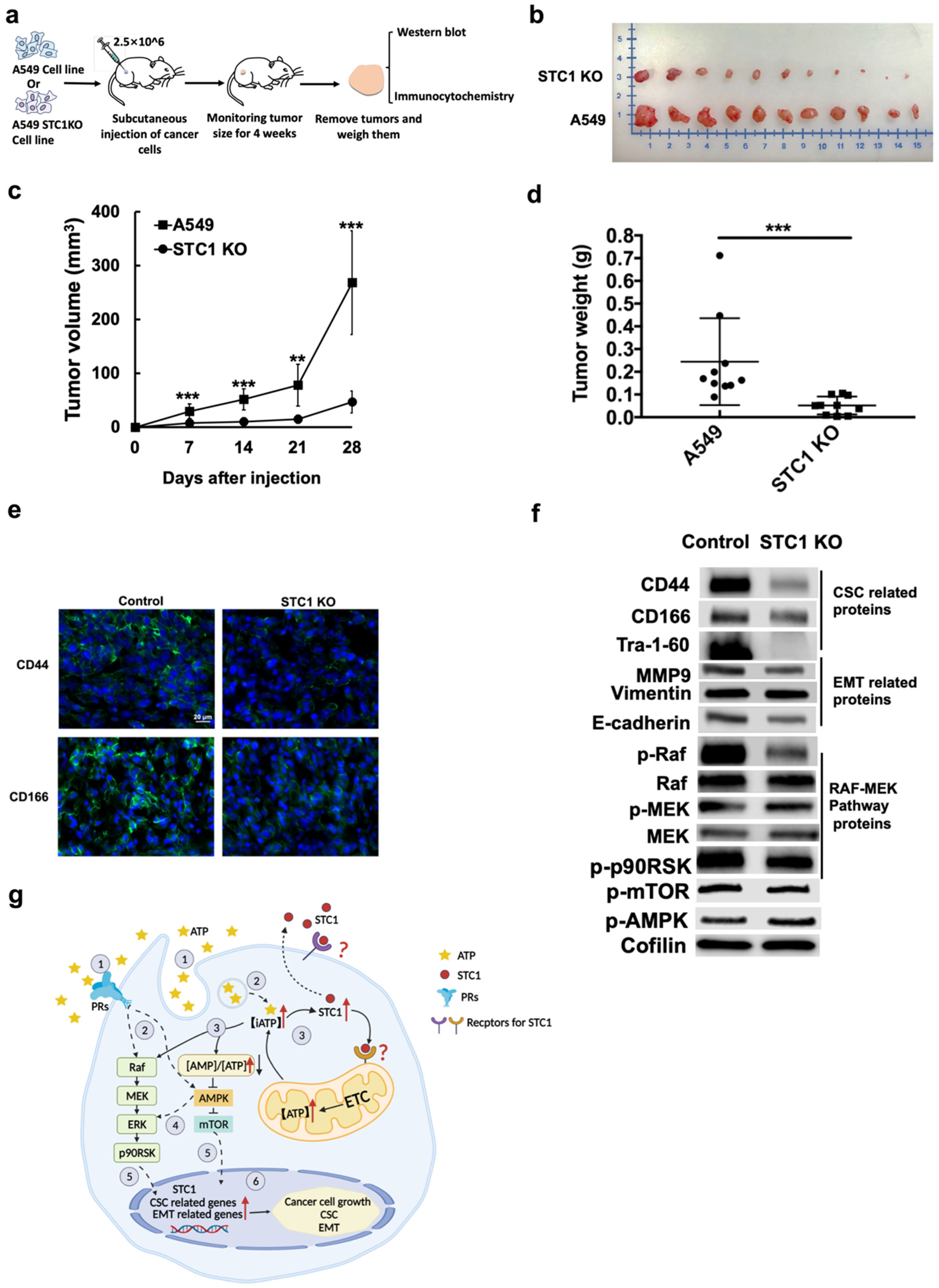
2.6. eATP-Treated A549 Cells Formed More Tumors at Lower Cell Injection Numbers, and Tumors Formed from A549stc1ko Cells Grew Slower and With Fewer CSCs
2.6.1. First Animal Study
2.6.2. Second Animal Study
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Transwell Migration and Invasion Assays
4.3. qRT-PCR
4.4. Colony Formation Assay
4.5. Western Blot Analysis
4.6. Immunofluorescence Microscopy
4.7. Flow Cytometry/Cell Sorter Assay
4.8. RNA Sequencing
4.9. Cell Proliferation and ATP Assays
4.10. STC1 Gene Knockdown (KD) and Knockout (KO)
4.11. Cell Growth Curve
4.12. Seahorse Metabolic Studies
4.13. Tumor Studies
4.14. Immunocytochemistry Study of Tumor Sections
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer stem cells: Significance in origin, pathogenesis, and treatment of glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, C.; Weinberg, R.A. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells. Int. J. Cancer 2011, 129, 2310–2314. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lee, S.O.; Xia, S.; Jiang, Q.; Luo, J.; Li, L.; Yeh, S.; Chang, C. Endothelial cells enhance prostate cancer metastasis via IL-6→androgen receptor→TGF-β→MMP-9 signals. Mol. Cancer Ther. 2013, 12, 1026–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Falzoni, S.; Donvito, G.; Di Virgilio, F. Detecting adenosine triphosphate in the pericellular space. Interface Focus 2013, 3, 20120101. [Google Scholar] [CrossRef] [Green Version]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (New York N. Y.) 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, K.; Ganesan, J.; Müller, T.; Dürr, C.; Grimm, M.; Beilhack, A.; Krempl, C.D.; Sorichter, S.; Gerlach, U.V.; Jüttner, E.; et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 2010, 16, 1434–1438. [Google Scholar] [CrossRef]
- Dubyak, G.R.; el-Moatassim, C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am. J. Physiol. 1993, 265 Pt 1, C577–C606. [Google Scholar] [CrossRef] [Green Version]
- Schenk, U.; Westendorf, A.M.; Radaelli, E.; Casati, A.; Ferro, M.; Fumagalli, M.; Verderio, C.; Buer, J.; Scanziani, E.; Grassi, F. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci. Signal. 2008, 1, ra6. [Google Scholar] [CrossRef] [Green Version]
- de Andrade Mello, P.; Coutinho-Silva, R.; Savio, L. Multifaceted effects of extracellular adenosine triphosphate and adenosine in the tumor-host interaction and therapeutic perspectives. Front. Immunol. 2017, 8, 1526. [Google Scholar] [CrossRef] [Green Version]
- Grygorczyk, R.; Boudreault, F.; Ponomarchuk, O.; Tan, J.J.; Furuya, K.; Goldgewicht, J.; Kenfack, F.D.; Yu, F. Lytic release of cellular ATP: Physiological relevance and therapeutic applications. Life 2021, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dourado, M.; Wong, E.; Hackos, D.H. Pannexin-1 is blocked by its C-terminus through a delocalized non-specific interaction surface. PLoS ONE 2014, 9, e99596. [Google Scholar] [CrossRef]
- Gaitán-Peñas, H.; Gradogna, A.; Laparra-Cuervo, L.; Solsona, C.; Fernández-Dueñas, V.; Barrallo-Gimeno, A.; Ciruela, F.; Lakadamyali, M.; Pusch, M.; Estévez, R. Investigation of LRRC8-mediated volume-regulated anion currents in Xenopus oocytes. Biophys. J. 2016, 111, 1429–1443. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, Y.; Hiasa, M.; Sakamoto, S.; Omote, H.; Nomura, M. Vesicular nucleotide transporter (VNUT): Appearance of an actress on the stage of purinergic signaling. Purinergic Signal. 2017, 13, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.A. Connexin and Pannexin hemichannels are regulated by redox potential. Front. Physiol. 2014, 5, 80. [Google Scholar] [CrossRef] [Green Version]
- Sandilos, J.K.; Chiu, Y.H.; Chekeni, F.B.; Armstrong, A.J.; Walk, S.F.; Ravichandran, K.S.; Bayliss, D.A. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J. Biol. Chem. 2012, 287, 11303–11311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Ramírez, A.S.; Díaz-Muñoz, M.; Butanda-Ochoa, A.; Vázquez-Cuevas, F.G. Nucleotides and nucleoside signaling in the regulation of the epithelium to mesenchymal transition (EMT). Purinergic Signal. 2017, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, X.; Li, Y.; Evers, M.; Zhang, H.; Chen, X. Extracellular and macropinocytosis internalized ATP work together to induce epithelial-mesenchymal transition and other early metastatic activities in lung cancer. Cancer Cell Int. 2019, 19, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Wang, X.; Li, Y.; Cao, Y.; Chen, X. Extracellular ATP a new player in cancer metabolism: NSCLC cells internalize ATP in vitro and in vivo using multiple endocytic mechanisms. Mol. Cancer Res. 2016, 14, 1087–1096. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Wang, X.; Liu, Y.; Li, Y.; Colvin, R.A.; Tong, L.; Wu, S.; Chen, X. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett. 2014, 351, 242–251. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Qian, Y.; Cao, Y.; Shriwas, P.; Zhang, H.; Chen, X. Extracellular ATP, as an energy and phosphorylating molecule, induces different types of drug resistances in cancer cells through ATP internalization and intracellular ATP level increase. Oncotarget 2017, 8, 87860–87877. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance (Alhambra Calif.) 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, C.M.; Qian, Y.; Adhicary, S.; Li, Y.; Shriwas, P.; Wang, X.; Bachmann, L.; Chen, X. Fluorescence microscopy for ATP internalization mediated by macropinocytosis in human tumor cells and tumor-xenografted mice. J. Vis. Exp. 2021, 30, e62768. [Google Scholar] [CrossRef] [PubMed]
- Commisso, C. The pervasiveness of macropinocytosis in oncological malignancies. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2019, 374, 20180153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, J.A.; King, J.S. The breadth of macropinocytosis research. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2019, 374, 20180146. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Kalra, H.; Akundi, R.S. Extracellular ATP mediates cancer cell migration and invasion through increased expression of cyclooxygenase 2. Front. Pharmacol. 2021, 11, 617211. [Google Scholar] [CrossRef]
- Evers, M.; Song, J.; Shriwas, P.; Greenbaum, H.S.; Chen, X. From transcriptomics, metabolomics to functional studies: Extracellular ATP induces TGF-β-like epithelial mesenchymal transition in lung cancer cells. Front. Oncol. 2022, 2948. [Google Scholar] [CrossRef]
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Cancer Facts & Figures 2021; American Cancer Society: Atlanta, GA, USA, 2021. [Google Scholar]
- Debieuvre, D.; Molinier, O.; Falchero, L.; Locher, C.; Templement-Grangerat, D.; Meyer, N.; Morel, H.; Duval, Y.; Asselain, B.; Letierce, A.; et al. Study Group KBP-2020-CPHG, & KBP-2020-CPHG Lung cancer trends and tumor characteristic changes over 20 years (2000–2020): Results of three French consecutive nationwide prospective cohorts’ studies. Lancet Reg. Health Eur. 2022, 22, 100492. [Google Scholar] [CrossRef]
- Buhrmann, C.; Yazdi, M.; Popper, B.; Kunnumakkara, A.B.; Aggarwal, B.B.; Shakibaei, M. Induction of the epithelial-to-mesenchymal transition of human colorectal cancer by human TNF-β (Lymphotoxin) and its reversal by resveratrol. Nutrients 2019, 11, 704. [Google Scholar] [CrossRef]
- Buhrmann, C.; Kraehe, P.; Lueders, C.; Shayan, P.; Goel, A.; Shakibaei, M. Curcumin suppresses crosstalk between colon cancer stem cells and stromal fibroblasts in the tumor microenvironment: Potential role of EMT. PLoS ONE 2014, 9, e107514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCudden, C.R.; Kogon, M.R.; DiMattia, G.E.; Wagner, G.F. Novel expression of the stanniocalcin gene in fish. J. Endocrinol. 2001, 171, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; He, Z.C.; Zhang, X.N.; Liu, Q.; Chen, C.; Zhu, Z.; Chen, Q.; Shi, Y.; Yao, X.H.; Cui, Y.H.; et al. Stanniocalcin-1 augments stem-like traits of glioblastoma cells through binding and activating NOTCH1. Cancer Lett. 2018, 416, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Ellard, J.P.; McCudden, C.R.; Tanega, C.; James, K.A.; Ratkovic, S.; Staples, J.F.; Wagner, G.F. The respiratory effects of stanniocalcin-1 (STC-1) on intact mitochondria and cells: STC-1 uncouples oxidative phosphorylation and its actions are modulated by nucleotide triphosphates. Mol. Cell. Endocrinol. 2007, 264, 90–101. [Google Scholar] [CrossRef]
- Zhang, K.Z.; Lindsberg, P.J.; Tatlisumak, T.; Kaste, M.; Olsen, H.S.; Andersson, L.C. Stanniocalcin: A molecular guard of neurons during cerebral ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 3637–3642. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Takeda, T.; Michiue, H.; Abe, T.; Takei, K. Actin bundling by dynamin 2 and cortactin is implicated in cell migration by stabilizing filopodia in human non-small cell lung carcinoma cells. Int. J. Oncol. 2016, 49, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95 (Suppl. 1), S20–S25. [Google Scholar] [CrossRef]
- Zhu, P.; Fan, Z. Cancer stem cells and tumorigenesis. Biophys. Rep. 2018, 4, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer stem cells: The architects of the tumor ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.C.; Doherty, J.; Huschtscha, L.I.; Redvers, R.; Restall, C.; Reddel, R.R.; Anderson, R.L. STC1 expression is associated with tumor growth and metastasis in breast cancer. Clin. Exp. Metastasis 2015, 32, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.Z.; Gu, X.H.; Cheng, S.F.; Li, L.; Liu, H.; Hu, L.P.; Gao, F. The oncogenetic role of stanniocalcin 1 in lung adenocarcinoma: A promising serum candidate biomarker for tracking lung adenocarcinoma progression. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 5633–5644. [Google Scholar] [CrossRef]
- Fang, Z.; Tian, Z.; Luo, K.; Song, H.; Yi, J. Clinical significance of stanniocalcin expression in tissue and serum of gastric cancer patients. Chin. J. Cancer Res. (Chung-Kuo Yen Cheng Yen Chiu) 2014, 26, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lang, L.; He, L.; Gao, L.; Chyan, D.; Xiong, Y.; Li, H.; Peng, H.; Teng, Y. Intracellular reduction in ATP levels contributes to CYT997-induced suppression of metastasis of head and neck squamous carcinoma. J. Cell. Mol. Med. 2019, 23, 1174–1182. [Google Scholar] [CrossRef]
- Abe, K.; Kanehira, M.; Ohkouchi, S.; Kumata, S.; Suzuki, Y.; Oishi, H.; Noda, M.; Sakurada, A.; Miyauchi, E.; Fujiwara, T.; et al. Targeting stanniocalcin-1-expressing tumor cells elicits efficient antitumor effects in a mouse model of human lung cancer. Cancer Med. 2021, 10, 3085–3100. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Scimè, A. Metabolic regulation of epithelial to mesenchymal transition: Implications for endocrine cancer. Front. Endocrinol. 2019, 10, 773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Ózsvári, B.; Sotgia, F.; Lisanti, M.P. High ATP production fuels cancer drug resistance and metastasis: Implications for mitochondrial ATP depletion therapy. Front. Oncol. 2021, 11, 740720. [Google Scholar] [CrossRef]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic adaptations in cancer stem cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Schulien, I.; Hockenjos, B.; van Marck, V.; Ayata, C.K.; Follo, M.; Thimme, R.; Hasselblatt, P. Extracellular ATP and purinergic P2Y2 receptor signaling promote liver tumorigenesis in mice by exacerbating DNA damage. Cancer Res. 2020, 80, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Shriwas, P.; Roberts, D.; Li, Y.; Wang, L.; Qian, Y.; Bergmeier, S.; Hines, J.; Adhicary, S.; Nielsen, C.; Chen, X. A small-molecule pan-class I glucose transporter inhibitor reduces cancer cell proliferation in vitro and tumor growth in vivo by targeting glucose-based metabolism. Cancer Metab. 2021, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A feasible target for cancer therapy. Cells 2020, 9, 2496. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Log2(FC) Values | |||||
|---|---|---|---|---|---|
| Gene Symbol | Gene Name | ATP 2h | TGF-β 2h | ATP 6h | TGF-β 6h |
| 1. FoxO1 | Forkhead box O1 | 2.36 | 1.23 | 0.69 | 1.07 |
| 2. FOSL1 | FOS like 1, AP-1 transcription factor subunit | 2.44 | 0.58 | 1.24 | 0.14 |
| 3. FOS | Fos proto-oncogene, AP-1 transcription factor subunit | −4.20 | −3.21 | −1.05 | −3.32 |
| 4. JUNB | JunB proto-oncogene, AP-1 transcription factor subunit | −1.34 | 2.30 | −0.16 | 2.34 |
| 5. JUN | Jun proto-oncogene, AP-1 transcription factor subunit | 0.50 | 1.20 | 0.05 | 1.34 |
| 6. c-Maf/maf | MAF bZIP transcription factor | 0.81 | 3.54 | 0.80 | 4.75 |
| 7. MITF | Melanocyte inducing transcription factor | 1.10 | −1.00 | 0.16 | −1.16 |
| 8. NFkB1 | Nuclear factor kappa B subunit 1 | 1.49 | 1.13 | 0.05 | 0.97 |
| 9. SNAI1/Snail 1 | Snail family transcriptional repressor 1 | 0.21 | 2.85 | 0.50 | 3.13 |
| 10. SOX8 | SRY-box 8 | 2.01 | 2.72 | 2.94 | 2.91 |
| 11. SOX4 | SRY-box 4 | 0.62 | 1.28 | 0.10 | 0.93 |
| 12. SOX21 | SRY-box 21 | −0.51 | −2.00 | 0.28 | −2.91 |
| 13. SOX2 | SRY-box 2 | −1.90 | −2.90 | −0.73 | −3.39 |
| 14. Oct-3/4—POU5F1 | POU class 5 homeobox 1 | 0.31 | 0.85 | 0.76 | 0.94 |
| 15. Nanog | Nanog homeobox | −4.92 | 2.83 | −4.92 | −4.92 |
| Log2(FC) Values | |||||
|---|---|---|---|---|---|
| Gene Symbol | Gene Name | ATP 2h | TGF-β 2h | ATP 6h | TGF-β 6h |
| 1. BMP7 | Bone morphogenetic protein 7 | −0.09 | 1.36 | −0.66 | 0.69 |
| 2. E-Cadherin/CDH1 | Cadherin 1 | 0.23 | 0.04 | −0.82 | −1.17 |
| 3. LMO2 | LIM domain only 2 | 0.99 | 1.20 | 0.96 | 0.56 |
| 4. NOTCH1 | Notch 1 | −1.45 | 0.24 | 0.28 | 0.17 |
| 5. Sonic Hedgehog/SHH | Sonic hedgehog signaling molecule | −0.29 | 1.51 | −0.76 | 0.38 |
| 6. TRA-1-81/PODXL/TRA-1-60 | Podocalyxin like | −0.16 | 0.26 | 0.62 | 1.99 |
| 7. Vimentin/VIM | Vimentin | −0.10 | 0.36 | 0.18 | 1.06 |
| 8. CXCR4 | C-X-C motif chemokine receptor 4 | −1.19 | −1.02 | −0.21 | −0.87 |
| 9. IL6R | Interleukin 6 receptor | −0.10 | −0.23 | 1.12 | −2.30 |
| 10. Aminopeptidase N/CD13/ANPEP | Alanyl aminopeptidase, membrane | 0.35 | 0.58 | 0.42 | 1.09 |
| 11. CXCL8/IL-8 | C-X-C motif chemokine ligand 8 | 1.93 | 0.73 | 0.56 | 0.28 |
| 12. IL6 | Interleukin 6 | 2.10 | 0.99 | 0.60 | 2.08 |
| Number of Tumors Generated out of 10 Injections (X/10) | ||
|---|---|---|
| Number of Cells Injected | ATP Pretreatment | Without ATP Pretreatment |
| 3 × 106 | 10/10 | 10/10 |
| 1 × 106 | 10/10 | 9/10 |
| 3 × 105 | 9/10  8/10 8/10 | |
| 1 × 105 | 8/10  4/10 4/10 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Qian, Y.; Evers, M.; Nielsen, C.M.; Chen, X. Cancer Stem Cell Formation Induced and Regulated by Extracellular ATP and Stanniocalcin-1 in Human Lung Cancer Cells and Tumors. Int. J. Mol. Sci. 2022, 23, 14770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232314770
Song J, Qian Y, Evers M, Nielsen CM, Chen X. Cancer Stem Cell Formation Induced and Regulated by Extracellular ATP and Stanniocalcin-1 in Human Lung Cancer Cells and Tumors. International Journal of Molecular Sciences. 2022; 23(23):14770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232314770
Chicago/Turabian StyleSong, Jingwen, Yanrong Qian, Maria Evers, Corinne M. Nielsen, and Xiaozhuo Chen. 2022. "Cancer Stem Cell Formation Induced and Regulated by Extracellular ATP and Stanniocalcin-1 in Human Lung Cancer Cells and Tumors" International Journal of Molecular Sciences 23, no. 23: 14770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232314770