
Deletion of Notch3 Impairs Contractility of Renal Resistance Vessels Due to Deficient Ca2+ Entry
 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
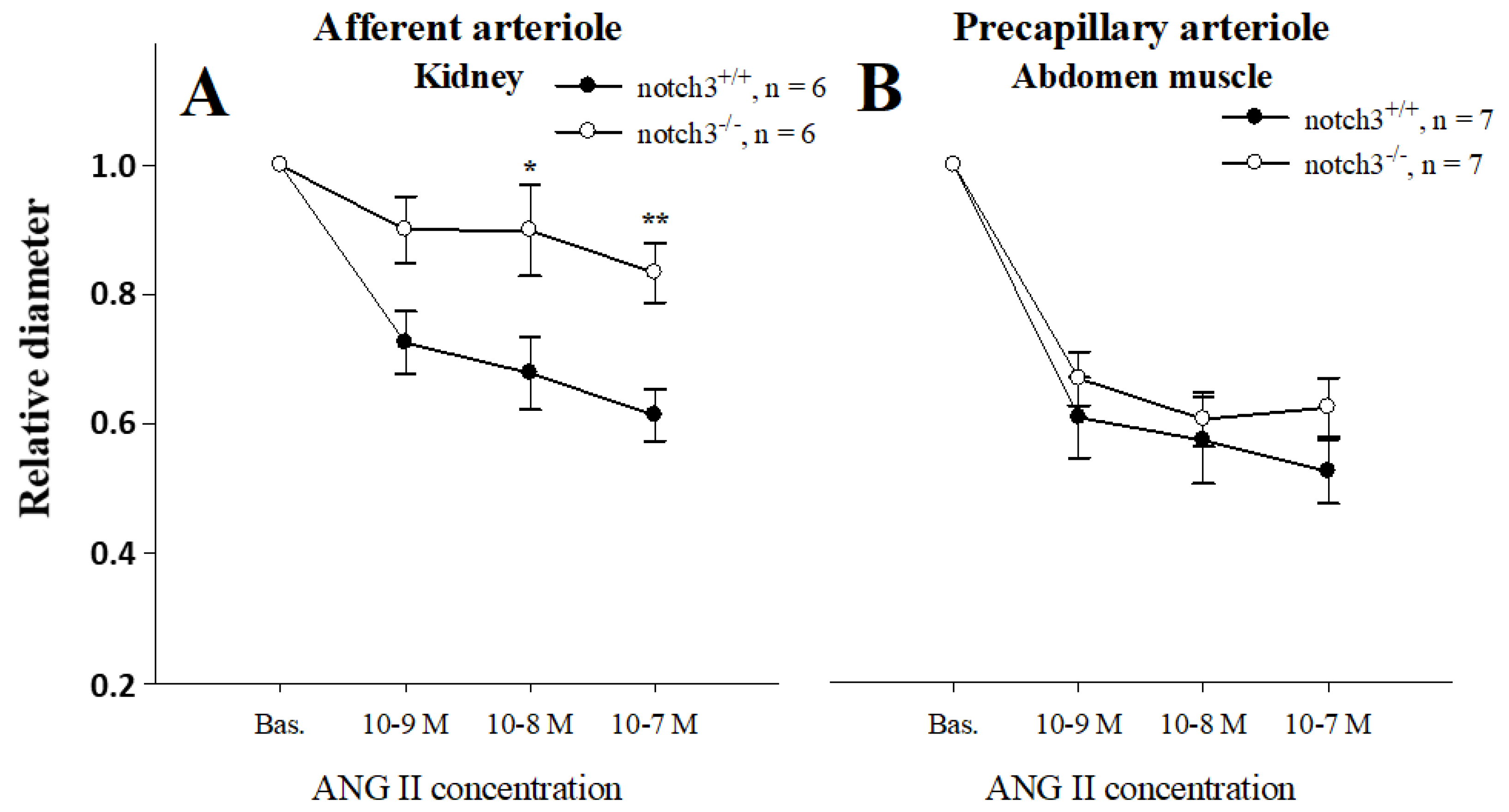
2.1. Notch3 Deficiency Affects Vascular Contractility in a Tissue-Specific Manner
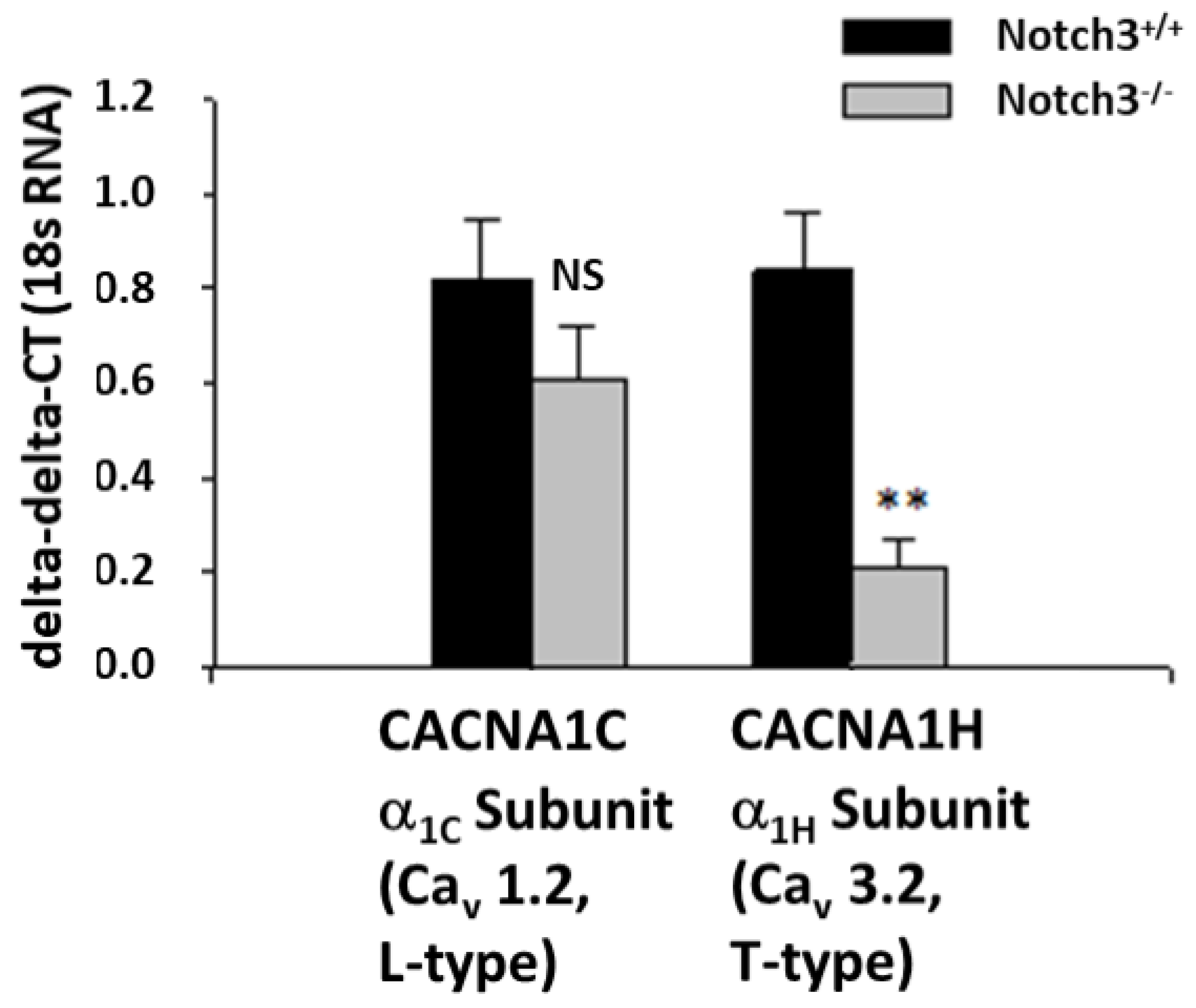
2.2. Notch3−/− Mice Display Decreased Levels of the T-Type Ca2+ Channel Subunit α1H in the Renal Cortex
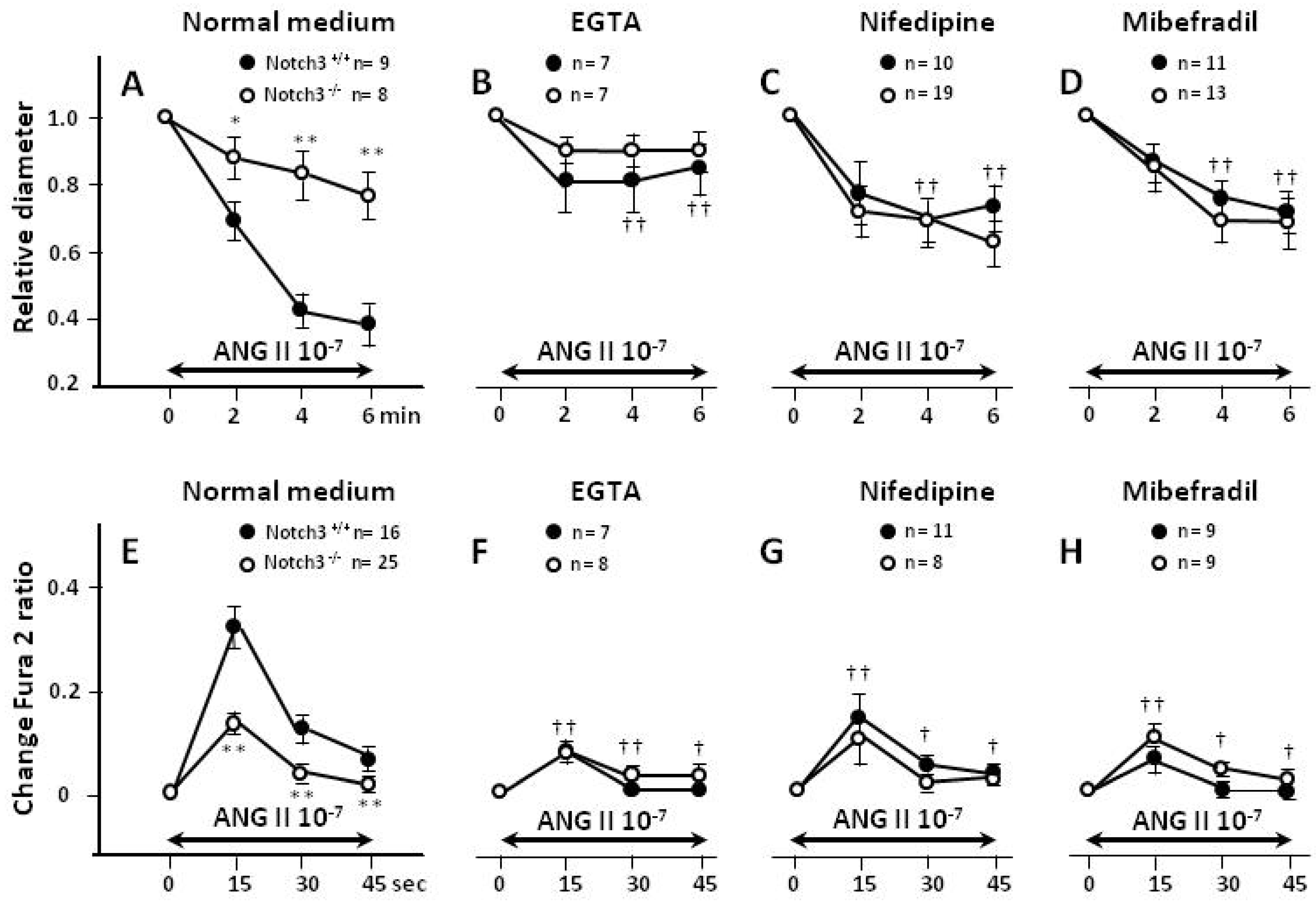
2.3. Blunted Contractile Responses of AAs from Notch3−/− Mice Are Associated with Decreased Intracellular Ca2+ (Ca2+i) Levels
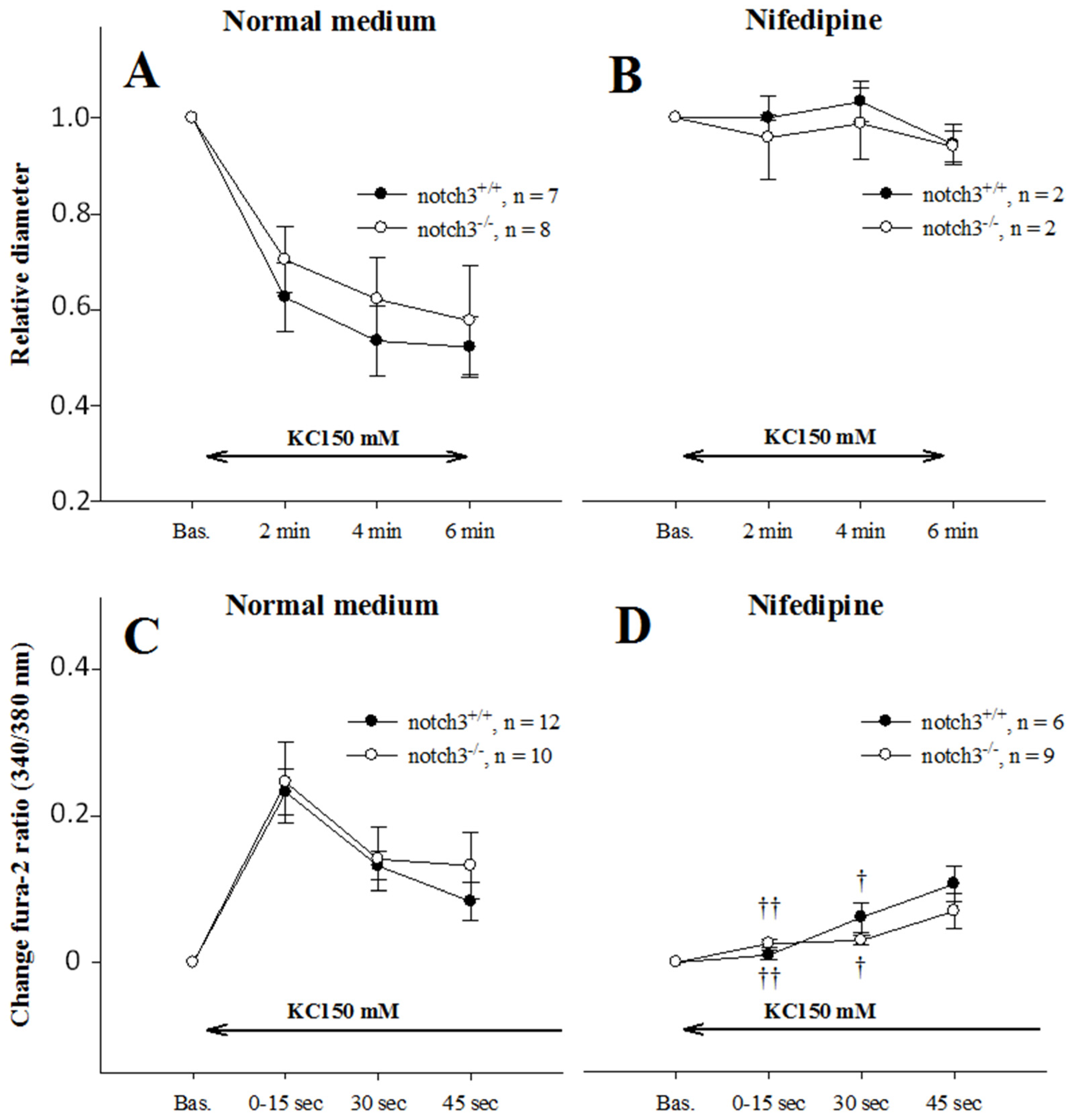
2.4. Extracellular Entry of Ca2+ Is Compromised in AAs from Notch3−/− Mice
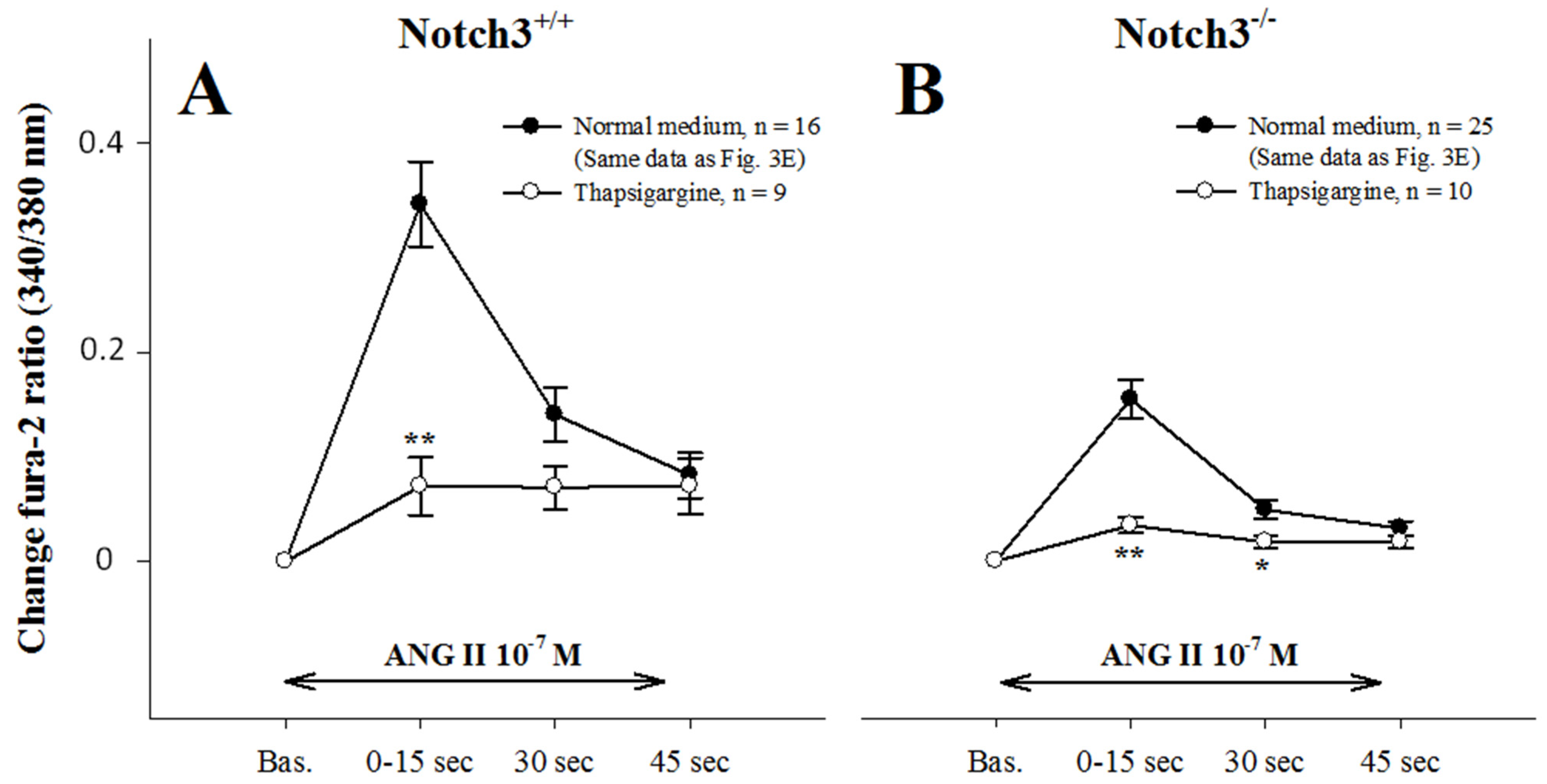
2.5. Intracellular Ca2+ Mobilization Is Normal in AAs from Notch3−/− Mice
2.6. AAs from Notch3−/− Mice Depolarize Normally
3. Discussion
4. Materials and Methods
4.1. Animals
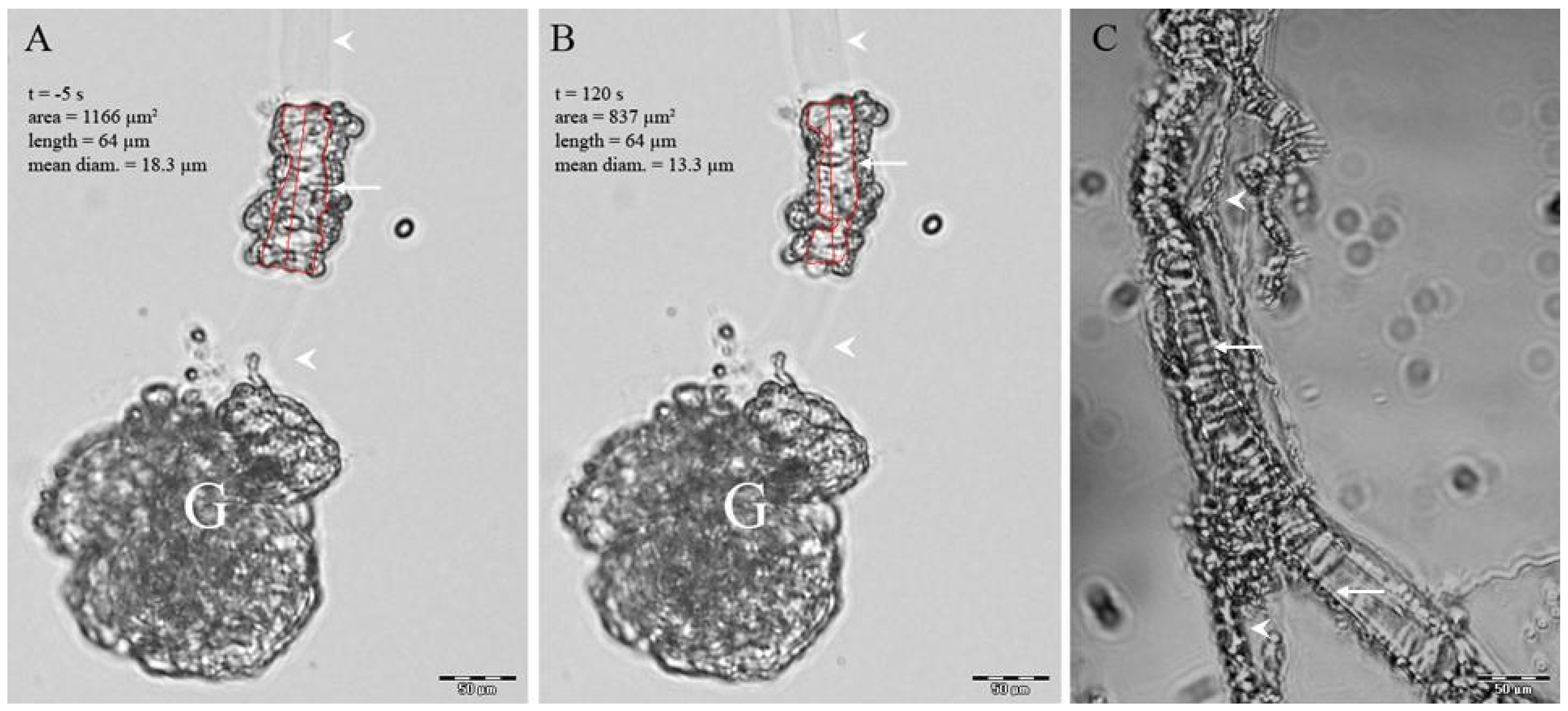
4.2. Isolation of Preglomerular Vessels
4.3. Measurement of Vessels Diameter and Ca2+i Responses
4.4. mRNA Extraction and Expression Analysis Using RT-PCR
4.5. Statistical Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Isolation of Preglomerular Vessels

Appendix A.2. Diameter and Fura-2 Ca2+i Responses to ANG II and KCl
Appendix A.3. Ca2+i Measurements
Appendix A.4. Chemicals-Reagents

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Gene | Symbol | ABI-Assay | WT (n = 11) | KO (n = 11) | Log2FC | p= |
|---|---|---|---|---|---|---|---|
| 1 | Notch-3 | Notch-3 | Notch3-Mm00435270_m1 | 1.55 ± 0.08 | 0.17 ± 0.16 | −3.18 | 0.0000004 |
| 2 | T-type Ca channel | Cacna1h | Cacna1h-Mm00445369_m1 | 0.84 ± 0.12 | 0.21 ± 0.06 | −2.02 | 0.0003 |
| 3 | Connexin 43 | Gja1 | Gja1-Mm00439105_m1 | 0.92 ± 0.13 | 0.51 ± 0.05 | −0.85 | 0.009 |
| 4 | smooth muscle actin | Acta2 | Acta2-Mm00725412_s1 | 0.91 ± 0.12 | 0.54 ± 0.06 | −0.75 | 0.01 |
| 5 | MYPT1 | Ppp1r12a | Ppp1r12a-Mm01234114_m1 | 0.60 ± 0.12 | 0.29 ± 0.04 | −1.06 | 0.02 |
| 6 | Transient receptor potential C6 | Trpc6 | Trpc6-Mm01176083_m1 | 1.27 ± 0.19 | 0.72 ± 0.11 | −0.81 | 0.02 |
| 7 | Hey2 | Hey2 | Hey2-Mm00469280_m1 | 0.76 ± 0.05 | 0.55 ± 0.07 | −0.46 | 0.03 |
| 8 | VE-cadherin | Cdh5 | Cdh5-Mm00486938_m1 | 0.74 ± 0.05 | 0.6 ± 0.04 | −0.3 | 0.03 |
| 9 | rhoA | Rhoa | Rhoa-Mm00834507_g1 | 0.69 ± 0.03 | 0.62 ± 0.02 | −0.15 | 0.04 |
| 10 | Intergin-linked kinase | Ilk | Ilk-Mm00439671_g1 | 0.90 ± 0.05 | 0.73 ± 0.05 | −0.3 | 0.04 |
| 11 | IP3-receptor | Ip3r1 | Itpr1-Mm00439917_m1 | 0.68 ± 0.02 | 0.62 ± 0.02 | −0.12 | 0.05 |
| 12 | PKC-alpha | prkca | Prkca-Mm00440858_m1 | 0.78 ± 0.03 | 0.68 ± 0.04 | −0.2 | 0.05 |
| 13 | calmodulin1 | CaM | Calm1-Mm00486655_m1 | 0.64 ± 0.03 | 0.57 ± 0.02 | −0.16 | 0.07 |
| 14 | MLCP-catalytic subunit beta | Ppp1cb | Ppp1cb-Mm00554690_m1 | 0.30 ± 0.02 | 0.24 ± 0.03 | −0.32 | 0.08 |
| 15 | DMPK | dmpk | Dmpk-Mm00446261_m1 | 0.81 ± 0.08 | 0.63 ± 0.05 | −0.35 | 0.08 |
| 16 | MLCK | Mylk | Mylk-Mm00653039_m1 | 0.72 ± 0.03 | 0.64 ± 0.04 | −0.17 | 0.1 |
| 17 | Cpi17 | Ppp1r14a | Ppp1r14a-Mm00471820_m1 | 0.88 ± 0.08 | 0.72 ± 0.05 | −0.29 | 0.11 |
| 18 | ADP-ribosyl-cyclase | Cd38 | Cd38-Mm00483146_m1 | 0.63 ± 0.03 | 0.56 ± 0.03 | −0.18 | 0.13 |
| 19 | Jag2 | Jag2 | Jag2-Mm00439935_m1 | 0.81 ± 0.06 | 0.68 ± 0.06 | −0.26 | 0.14 |
| 20 | ryanodine receptor 2 | ryr2 | Ryr2-Mm00465877_m1 | 1.13 ± 0.16 | 0.85 ± 0.1 | −0.42 | 0.14 |
| 21 | Connexin 37 | Gja4 | Gja4-Mm00433610_s1 | 0.85 ± 0.07 | 0.73 ± 0.05 | −0.22 | 0.16 |
| 22 | K+ inw rect channel J11 ATPsens | Kcnj11 | Kcnj11-Mm00440050_s1 | 1.21 ± 0.13 | 0.97 ± 0.11 | −0.32 | 0.16 |
| 23 | HeyL | HeyL | Heyl-Mm00516555_m1 | 0.79 ± 0.07 | 0.65 ± 0.07 | −0.28 | 0.17 |
| 24 | Tropomyosin 1 | Tpm1 | Tpm1-Mm00600378_m1 | 0.81 ± 0.09 | 0.67 ± 0.05 | −0.28 | 0.17 |
| 25 | ryanodine receptor 1 | ryr1 | Ryr1-Mm01175211_m1 | 1.35 ± 0.2 | 0.94 ± 0.21 | −0.53 | 0.17 |
| 26 | L-type Ca Channel | CACNA1C | Cacna1c-Mm00437917_m1 | 0.82 ± 0.13 | 0.61 ± 0.11 | −0.42 | 0.26 |
| 27 | Notch-4 | Notch-4 | Notch4-Mm00440536_g1 | 0.83 ± 0.05 | 0.77 ± 0.03 | −0.11 | 0.3 |
| 28 | Tbp | Tbp | Tbp-Mm00446973_m1 | 0.76 ± 0.02 | 0.72 ± 0.03 | −0.07 | 0.31 |
| 29 | Jag1 | Jag1 | Jag1-Mm00496902_m1 | 0.78 ± 0.06 | 0.7 ± 0.06 | −0.17 | 0.32 |
| 30 | connexin 40 | GJA5 | Gja5-Mm00433619_s1 | 0.66 ± 0.05 | 0.75 ± 0.08 | 0.19 | 0.33 |
| 31 | Notch-1 | Notch-1 | Notch1-Mm00435245_m1 | 0.84 ± 0.06 | 0.77 ± 0.06 | −0.12 | 0.39 |
| 32 | Raf-1 | Raf1 | Raf1-Mm00466513_m1 | 0.85 ± 0.04 | 0.8 ± 0.05 | −0.09 | 0.45 |
| 33 | P/Q-type Ca channel | CACNA1A | Cacna1a-Mm00432190_m1 | 1.01 ± 0.1 | 0.91 ± 0.12 | −0.16 | 0.49 |
| 34 | DLL1 | DLL1 | Dll1-Mm01279265_g1 | 0.64 ± 0.08 | 0.58 ± 0.04 | −0.14 | 0.55 |
| 35 | Hey1 | Hey1 | Hey1-Mm00468865_m1 | 0.96 ± 0.1 | 1.05 ± 0.12 | 0.13 | 0.57 |
| 36 | Ppia | Ppia | Ppia-Mm02342430_g1 | 0.79 ± 0.1 | 0.72 ± 0.08 | −0.14 | 0.58 |
| 37 | p38 MAPK | mapk14 | Mapk14-Mm00442497_m1 | 0.68 ± 0.03 | 0.66 ± 0.03 | −0.05 | 0.64 |
| 38 | MLCP-catalytic subunit alpha | Ppp1ca | Ppp1ca-Mm00453295_g1 | 0.58 ± 0.01 | 0.57 ± 0.02 | −0.03 | 0.69 |
| 39 | Cacn2d2 | Cacna2d2 | Cacna2d2-Mm00457825_m1 | 1.55 ± 0.24 | 1.66 ± 0.17 | 0.1 | 0.7 |
| 40 | MLCP-catalytic subunit gamma | Ppp1cc | Ppp1cc-Mm00849631_s1 | 0.84 ± 0.03 | 0.82 ± 0.04 | −0.03 | 0.71 |
| 41 | Zip Kinase, Dap-like-kinase | Dapk3 | Dapk3-Mm00492081_m1 | 0.69 ± 0.02 | 0.68 ± 0.03 | −0.02 | 0.72 |
| 42 | At1aR | Agtr1a | Agtr1a-Mm01166161_m1 | 0.65 ± 0.03 | 0.67 ± 0.04 | 0.03 | 0.76 |
| 43 | Hes1 | Hes1 | Hes1-Mm00468601_m1 | 0.64 ± 0.05 | 0.62 ± 0.06 | −0.04 | 0.82 |
| 44 | Notch-2 | Notch-2 | Notch2-Mm00803077_m1 | 0.77 ± 0.05 | 0.78 ± 0.05 | 0.01 | 0.96 |
| 45 | Pgk1 | Pgk1 | Pgk1-Mm00435617_m1 | 0.63 ± 0.02 | 0.63 ± 0.02 | 0 | 0.99 |
| 46 | rho-k | Grk1 | Grk1-Mm00442317_m1 | Not Detected | Not Detected | 0 | NA |
| 47 | Hes5 | Hes5 | Hes5-Mm00439311_g1 | Not Detected | Not Detected | 0 | NA |
References
- Iso, T.; Kedes, L.; Hamamori, Y. HES and HERP families: Multiple effectors of the Notch signaling pathway. J. Cell. Physiol. 2003, 194, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Schweisguth, F. Regulation of notch signaling activity. Curr. Biol. 2004, 14, R129–R138. [Google Scholar] [CrossRef] [PubMed]
- Villa, N.; Walker, L.; Lindsell, C.E.; Gasson, J.; Iruela-Arispe, M.L.; Weinmaster, G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech. Dev. 2001, 108, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Domenga, V.; Fardoux, P.; Lacombe, P.; Monet, M.; Maciazek, J.; Krebs, L.T.; Klonjkowski, B.; Berrou, E.; Mericskay, M.; Li, Z.; et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Gen Dev. 2004, 18, 2730–2735. [Google Scholar] [CrossRef] [Green Version]
- Belin de Chantemele, E.J.; Retailleau, K.; Pinaud, F.; Vessieres, E.; Bocquet, A.; Guihot, A.L.; Lemaire, B.; Domenga, V.; Baufreton, C.; Loufrani, L.; et al. Notch3 is a major regulator of vascular tone in cerebral and tail resistance arteries. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2216–2224. [Google Scholar] [CrossRef]
- Boulos, N.; Helle, F.; Dussaule, J.C.; Placier, S.; Milliez, P.; Djudjaj, S.; Guerrot, D.; Joutel, A.; Ronco, P.; Boffa, J.J.; et al. Notch3 is essential for regulation of the renal vascular tone. Hypertension 2011, 57, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Dichgans, M.; Mayer, M.; Uttner, I.; Bruning, R.; Muller-Hocker, J.; Rungger, G.; Ebke, M.; Klockgether, T.; Gasser, T. The phenotypic spectrum of CADASIL: Clinical findings in 102 cases. Ann. Neurol. 1998, 44, 731–739. [Google Scholar] [CrossRef]
- Tournier-Lasserve, E.; Joutel, A.; Melki, J.; Weissenbach, J.; Lathrop, G.M.; Chabriat, H.; Mas, J.L.; Cabanis, E.A.; Baudrimont, M.; Maciazek, J.; et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat. Gen. 1993, 3, 256–259. [Google Scholar] [CrossRef]
- Dubroca, C.; Lacombe, P.; Domenga, V.; Maciazek, J.; Levy, B.; Tournier-Lasserve, E.; Joutel, A.; Henrion, D. Impaired vascular mechanotransduction in a transgenic mouse model of CADASIL arteriopathy. Stroke 2005, 36, 113–117. [Google Scholar] [CrossRef]
- Ruchoux, M.M.; Domenga, V.; Brulin, P.; Maciazek, J.; Limol, S.; Tournier-Lasserve, E.; Joutel, A. Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am. J. Pathol. 2003, 162, 329–342. [Google Scholar] [CrossRef]
- Hansen, P.B.; Jensen, B.L.; Andreasen, D.; Skott, O. Differential expression of T- and L-type voltage-dependent calcium channels in renal resistance vessels. Circ. Res. 2001, 89, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Navar, L.G.; Gilmore, J.P.; Joyner, W.L.; Steinhausen, M.; Edwards, R.M.; Casellas, D.; Carmines, P.K.; Zimmerhackl, L.B.; Yokota, S.D. Direct assessment of renal microcirculatory dynamics. Fed. Proc. 1986, 45, 2851–2861. [Google Scholar] [PubMed]
- Guerrot, D.; Francois, A.; Boffa, J.J.; Boulos, N.; Hanoy, M.; Legallicier, B.; Triquenot-Bagan, A.; Guyant-Marechal, L.; Laquerriere, A.; Freguin-Bouilland, C.; et al. Nephroangiosclerosis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: Is NOTCH3 mutation the common culprit? Am. J. Kidney Dis. 2008, 52, 340–345. [Google Scholar] [CrossRef]
- Kusaba, T.; Hatta, T.; Kimura, T.; Sonomura, K.; Tanda, S.; Kishimoto, N.; Kameyama, H.; Okigaki, M.; Mori, Y.; Ishigami, N.; et al. Renal involvement in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Clin. Nephrol. 2007, 67, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Kaßmann, M.; Hashad, A.M.; Welsh, D.G.; Gollasch, M. Differential targeting and signaling of voltage-gated T-type Cav3.2 and L-type Cav1.2 channels to ryanodine receptors in mesenteric arteries. J. Physiol. 2018, 596, 4863–4877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellner, S.K.; Arendschorst, W.J. Voltage-gated Ca2+ entry and ryanodine receptor Ca2+-induced Ca2+ release in preglomerular arterioles. Am. J. Physiol. Ren. Physiol. 2007, 292, F1568–F1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Paez-Cortez, J.R.; Boppidi, K.; Vasconcelos, M.; Roy, M.; Cardoso, W.; Ai, X.; Fine, A. Activation dynamics and signaling properties of Notch3 receptor in the developing pulmonary artery. J. Biol. Chem. 2011, 286, 22678–22687. [Google Scholar] [CrossRef] [Green Version]
- Fellner, S.K.; Arendshorst, W.J. Angiotensin II Ca2+ signaling in rat afferent arterioles: Stimulation of cyclic ADP ribose and IP3 pathways. Am. J. Physiol. Ren. Physiol. 2005, 288, F785–F791. [Google Scholar] [CrossRef]
- Hayashi, K.; Wakino, S.; Homma, K.; Sugano, N.; Saruta, T. Pathophysiological significance of T-type Ca2+ channels: Role of T-type Ca2+ channels in renal microcirculation. J. Pharmacol. Sci. 2005, 99, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Perez-Reyes, E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef]
- Moosmang, S.; Haider, N.; Bruderl, B.; Welling, A.; Hofmann, F. Antihypertensive effects of the putative T-type calcium channel antagonist mibefradil are mediated by the L-type calcium channel Cav1.2. Circ. Res. 2006, 98, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.G.; Li, M.; Navar, L.G. T-type calcium channels in the regulation of afferent and efferent arterioles in rats. Am. J. Physiol. Ren. Physiol. 2004, 286, F331–F337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, P.A.; Yang, X.; Moss, N.G.; Arendshort, W.J. Superoxide enhances Ca2+ entry through L-type channels in the renal afferent arteriole. Hypertension 2015, 66, 374–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmines, P.K.; Fowler, B.C.; Bell, P.D. Segmentally distinct effects of depolarization on intracellular [Ca2+] in renal arterioles. Am. J. Physiol. Ren. Physiol. 1993, 265, F677–F685. [Google Scholar] [CrossRef] [PubMed]
- Loutzenhiser, R.; Hayashi, K.; Epstein, M. Divergent effects of KCl-induced depolarization on afferent and efferent arterioles. Am. J. Physiol. Ren. Physiol. 1989, 257, F561–F564. [Google Scholar] [CrossRef]
- Salomonsson, M.; Sorensen, C.M.; Arendshorst, W.J.; Steendahl, J.; Holstein-Rathlou, N.H. Calcium handling in afferent arterioles Acta Physiol. Scand. 2004, 181, 421–429. [Google Scholar]
- Tiemann, K.; Weyer, D.; Djoufack, P.C.; Ghanem, A.; Lewalter, T.; Dreiner, U.; Meyer, R.; Grohe, C.; Fink, K.B. Increasing myocardial contraction and blood pressure in C57BL/6 mice during early postnatal development. Am. J. Physiol. Heart Physiol. 2003, 284, H464–H474. [Google Scholar] [CrossRef] [Green Version]
- Arboleda-Velasquez, J.F.; Zhou, Z.; Shin, H.K.; Louvi, A.; Kim, H.H.; Savitz, S.I.; Liao, J.K.; Salomone, S.; Ayata, C.; Moskowitz, M.A.; et al. Linking Notch signaling to ischemic stroke. Proc. Natl. Acad. Sci. USA 2008, 105, 4856–4861. [Google Scholar] [CrossRef] [Green Version]
- Fellner, S.K.; Arendshorst, W.J. Angiotensin II-stimulated Ca2+ entry mechanisms in afferent arterioles: Role of transient receptor potential canonical channels and reverse Na+/Ca2+ exchange. Am. J. Physiol. Ren. Physiol. 2008, 294, F212–F219. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.P.; Sutherland, C.; Borman, M.A.; Deng, J.T.; Macdonald, J.A.; Walsh, M.P. Integrin-linked kinase is responsible for Ca2+-independent myosin diphosphorylation and contraction of vascular smooth muscle. Biochem. J. 2005, 392, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Loutzenhiser, K.; Loutzenhiser, R. Angiotensin II-induced Ca2+ influx in renal afferent and efferent arterioles: Differing roles of voltage-gated and store-operated Ca2+ entry. Circ. Res. 2000, 87, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, L.L. T-type Ca2+ channels in vascular smooth muscle: Multiple functions. Cell Calcium 2006, 40, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Holstein-Rathlou, N.H. Is there a role for T-type Ca2+ channels in regulation of vasomotor tone in mesenteric arterioles? Can. J. Physiol. Pharmacol. 2009, 87, 8–20. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helle, F.; Hultström, M.; Kavvadas, P.; Iversen, B.; Chadjichristos, C.E.; Chatziantoniou, C. Deletion of Notch3 Impairs Contractility of Renal Resistance Vessels Due to Deficient Ca2+ Entry. Int. J. Mol. Sci. 2022, 23, 16068. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232416068
Helle F, Hultström M, Kavvadas P, Iversen B, Chadjichristos CE, Chatziantoniou C. Deletion of Notch3 Impairs Contractility of Renal Resistance Vessels Due to Deficient Ca2+ Entry. International Journal of Molecular Sciences. 2022; 23(24):16068. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232416068
Chicago/Turabian StyleHelle, Frank, Michael Hultström, Panagiotis Kavvadas, Bjarne Iversen, Christos E. Chadjichristos, and Christos Chatziantoniou. 2022. "Deletion of Notch3 Impairs Contractility of Renal Resistance Vessels Due to Deficient Ca2+ Entry" International Journal of Molecular Sciences 23, no. 24: 16068. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232416068