
Emerging Insights on the Diverse Roles of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Chronic Liver Diseases: Cholesterol Metabolism and Beyond


Abstract
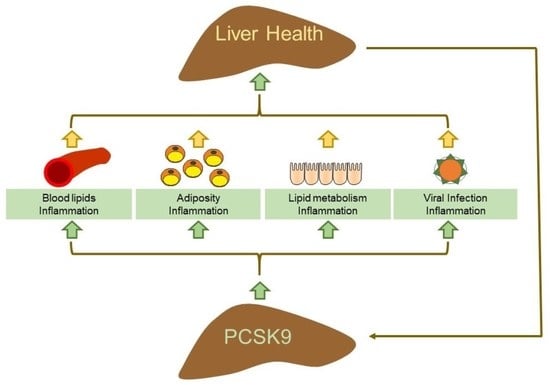
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
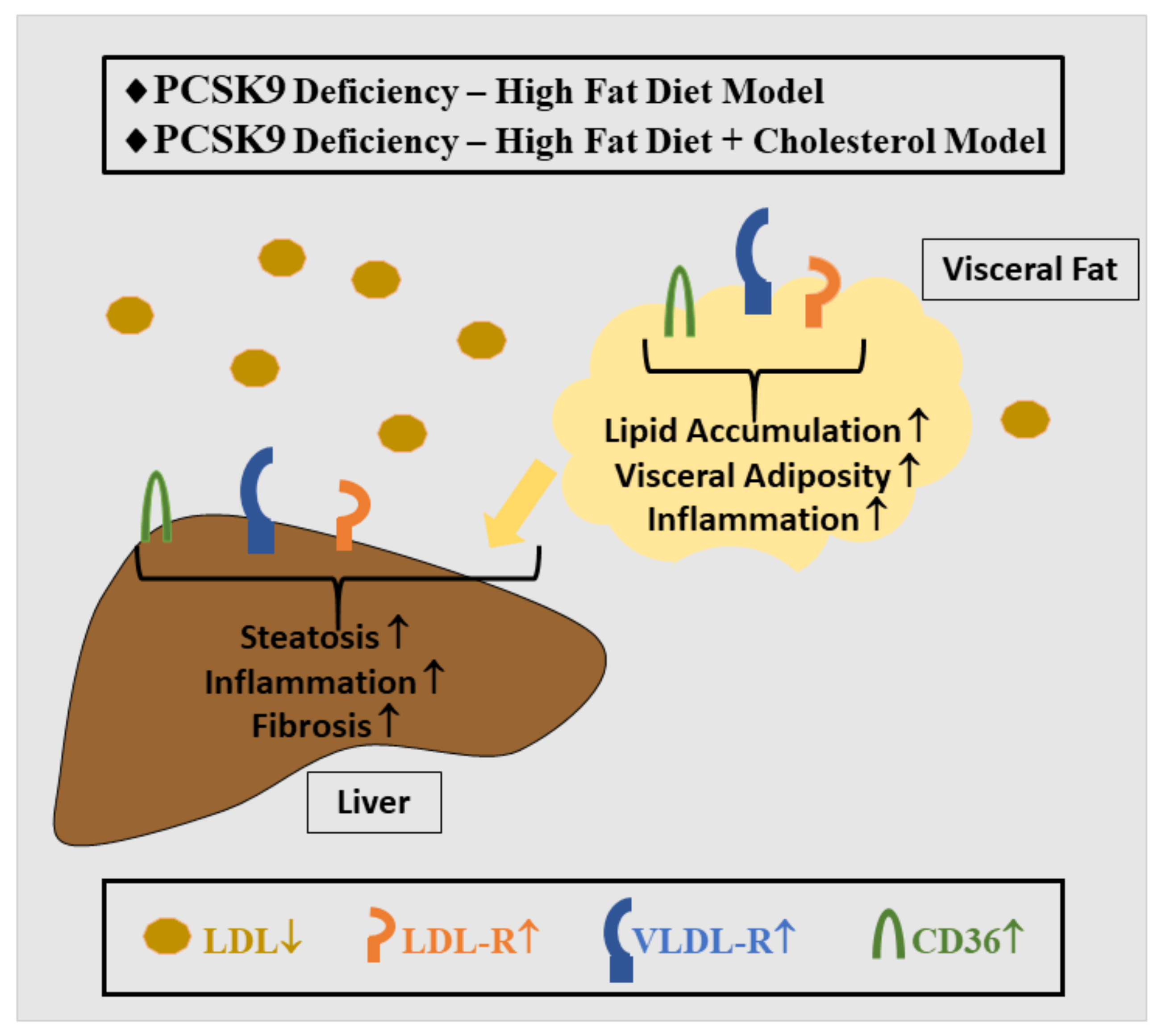{kind=link}
{kind=link}
1. Introduction
2. Roles for PCSK9 in the Intestine
3. Circulating PCSK9 and LDL-Cholesterol Levels in Different Patient Cohorts
4. Regulation of Hepatocyte PCSK9 by Cytokines and Adipokines
5. PCSK9 and Inflammation
6. PCSK9 in Patients with Liver Cirrhosis and Mixed Disease Etiology
7. PCSK9 and Alcoholic Liver Disease
8. PCSK9 and HCV-Induced Chronic Liver Disease

9. PCSK9 and NAFLD Pathogenesis
10. PCSK9 and Other Etiologies of Liver Diseases
11. PCSK9 and HCC Progression
12. Expression and Role of PCSK9 in Adipose Tissues
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Benjannet, S.; Rhainds, D.; Essalmani, R.; Mayne, J.; Wickham, L.; Jin, W.; Asselin, M.C.; Hamelin, J.; Varret, M.; Allard, D.; et al. NARC-1/PCSK9 and its natural mutants: Zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 2004, 279, 48865–48875. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, D.; Danley, D.E.; Geoghegan, K.F.; Griffor, M.C.; Hawkins, J.L.; Subashi, T.A.; Varghese, A.H.; Ammirati, M.J.; Culp, J.S.; Hoth, L.R.; et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 2007, 14, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Sato, R. SREBPs: Protein interaction and SREBPs. FEBS J. 2009, 276, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Moon, Y.A.; Horton, J.D. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 2004, 279, 50630–50638. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef]
- Sundararaman, S.S.; Doring, Y.; van der Vorst, E.P.C. PCSK9: A Multi-Faceted Protein That Is Involved in Cardiovascular Biology. Biomedicines 2021, 9, 793. [Google Scholar] [CrossRef] [PubMed]
- Nozue, T. Lipid Lowering Therapy and Circulating PCSK9 Concentration. J. Atheroscler. Thromb. 2017, 24, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Abifadel, M.; Varret, M.; Rabes, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Allard, D.; Amsellem, S.; Abifadel, M.; Trillard, M.; Devillers, M.; Luc, G.; Krempf, M.; Reznik, Y.; Girardet, J.P.; Fredenrich, A.; et al. Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Hum. Mutat. 2005, 26, 497. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Schulz, R.; Schluter, K.D. PCSK9 targets important for lipid metabolism. Clin. Res. Cardiol. Suppl. 2017, 12, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Essalmani, R.; Susan-Resiga, D.; Chamberland, A.; Abifadel, M.; Creemers, J.W.; Boileau, C.; Seidah, N.G.; Prat, A. In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 2011, 286, 4257–4263. [Google Scholar] [CrossRef] [Green Version]
- Cameron, J.; Holla, O.L.; Laerdahl, J.K.; Kulseth, M.A.; Ranheim, T.; Rognes, T.; Berge, K.E.; Leren, T.P. Characterization of novel mutations in the catalytic domain of the PCSK9 gene. J. Intern. Med. 2008, 263, 420–431. [Google Scholar] [CrossRef]
- Benjannet, S.; Rhainds, D.; Hamelin, J.; Nassoury, N.; Seidah, N.G. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: Functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 2006, 281, 30561–30572. [Google Scholar] [CrossRef] [Green Version]
- Zaid, A.; Roubtsova, A.; Essalmani, R.; Marcinkiewicz, J.; Chamberland, A.; Hamelin, J.; Tremblay, M.; Jacques, H.; Jin, W.; Davignon, J.; et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): Hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 2008, 48, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Lipari, M.T.; Li, W.; Moran, P.; Kong-Beltran, M.; Sai, T.; Lai, J.; Lin, S.J.; Kolumam, G.; Zavala-Solorio, J.; Izrael-Tomasevic, A.; et al. Furin-cleaved proprotein convertase subtilisin/kexin type 9 (PCSK9) is active and modulates low density lipoprotein receptor and serum cholesterol levels. J. Biol. Chem. 2012, 287, 43482–43491. [Google Scholar] [CrossRef] [Green Version]
- Oleaga, C.; Hay, J.; Gurcan, E.; David, L.L.; Mueller, P.A.; Tavori, H.; Shapiro, M.D.; Pamir, N.; Fazio, S. Insights into the kinetics and dynamics of the furin-cleaved form of PCSK9. J. Lipid Res. 2020, 62, 100003. [Google Scholar] [CrossRef]
- Caselli, C.; Del Turco, S.; Ragusa, R.; Lorenzoni, V.; De Graaf, M.; Basta, G.; Scholte, A.; De Caterina, R.; Neglia, D. Association of PCSK9 plasma levels with metabolic patterns and coronary atherosclerosis in patients with stable angina. Cardiovasc. Diabetol. 2019, 18, 144. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Skill, N.; Marcus, V.; Deschenes, M.; Tan, X.; Bouteaud, J.; Negi, S.; Awan, Z.; Aikin, R.; Kwan, J.; et al. Decreased PCSK9 expression in human hepatocellular carcinoma. BMC Gastroenterol. 2015, 15, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, E.; Cho, N.H.; Moon, S.S.; Cho, H. Comparison of Serum PCSK9 Levels in Subjects with Normoglycemia, Impaired Fasting Glucose, and Impaired Glucose Tolerance. Endocrinol. Metab. 2020, 35, 480–483. [Google Scholar] [CrossRef]
- Liu, J.; Guo, Y.L.; Xu, R.X.; Li, J.J. Rapid effects of different lipid-lowering drugs on PCSK9 in humans. Clin. Lipidol. 2013, 8, 519–524. [Google Scholar] [CrossRef]
- Cesaro, A.; Bianconi, V.; Gragnano, F.; Moscarella, E.; Fimiani, F.; Monda, E.; Scudiero, O.; Limongelli, G.; Pirro, M.; Calabro, P. Beyond cholesterol metabolism: The pleiotropic effects of proprotein convertase subtilisin/kexin type 9 (PCSK9). Genetics, mutations, expression, and perspective for long-term inhibition. Biofactors 2020, 46, 367–380. [Google Scholar] [CrossRef]
- Schlegel, V.; Treuner-Kaueroff, T.; Seehofer, D.; Berg, T.; Becker, S.; Ceglarek, U.; Thiery, J.; Kaiser, T. Low PCSK9 levels are correlated with mortality in patients with end-stage liver disease. PLoS ONE 2017, 12, e0181540. [Google Scholar] [CrossRef] [Green Version]
- Persson, L.; Cao, G.; Stahle, L.; Sjoberg, B.G.; Troutt, J.S.; Konrad, R.J.; Galman, C.; Wallen, H.; Eriksson, M.; Hafstrom, I.; et al. Circulating proprotein convertase subtilisin kexin type 9 has a diurnal rhythm synchronous with cholesterol synthesis and is reduced by fasting in humans. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2666–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, T.A. Diurnal variation of cholesterol precursors squalene and methyl sterols in human plasma lipoproteins. J. Lipid Res. 1982, 23, 466–473. [Google Scholar] [CrossRef]
- Ooi, T.C.; Krysa, J.A.; Chaker, S.; Abujrad, H.; Mayne, J.; Henry, K.; Cousins, M.; Raymond, A.; Favreau, C.; Taljaard, M.; et al. The Effect of PCSK9 Loss-of-Function Variants on the Postprandial Lipid and ApoB-Lipoprotein Response. J. Clin. Endocrinol. Metab. 2017, 102, 3452–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, N.; Ruscica, M.; Coggi, D.; Bonomi, A.; Amato, M.; Frigerio, B.; Sansaro, D.; Ravani, A.; Veglia, F.; Capra, N.; et al. Sex-specific predictors of PCSK9 levels in a European population: The IMPROVE study. Atherosclerosis 2020, 309, 39–46. [Google Scholar] [CrossRef]
- Grimaudo, S.; Bartesaghi, S.; Rametta, R.; Marra, F.; Margherita Mancina, R.; Pihlajamaki, J.; Kakol-Palm, D.; Andreasson, A.C.; Dongiovanni, P.; Ludovica Fracanzani, A.; et al. PCSK9 rs11591147 R46L loss-of-function variant protects against liver damage in individuals with NAFLD. Liver Int. 2021, 41, 321–332. [Google Scholar] [CrossRef]
- Grefhorst, A.; McNutt, M.C.; Lagace, T.A.; Horton, J.D. Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J. Lipid Res. 2008, 49, 1303–1311. [Google Scholar] [CrossRef] [Green Version]
- Feder, S.; Wiest, R.; Weiss, T.S.; Aslanidis, C.; Schacherer, D.; Krautbauer, S.; Liebisch, G.; Buechler, C. Proprotein convertase subtilisin/kexin type 9 (PCSK9) levels are not associated with severity of liver disease and are inversely related to cholesterol in a cohort of thirty eight patients with liver cirrhosis. Lipids Health Dis. 2021, 20, 6. [Google Scholar] [CrossRef]
- Parhofer, K.G.; von Stritzky, B.; Pietschmann, N.; Dorn, C.; Paar, W.D. PEARL: A Non-interventional Study of Real-World Alirocumab Use in German Clinical Practice. Drugs Real World Outcomes 2019, 6, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Katzmann, J.L.; Gouni-Berthold, I.; Laufs, U. PCSK9 Inhibition: Insights From Clinical Trials and Future Prospects. Front. Physiol. 2020, 11, 595819. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, G.; Zinzi, A.; Scavone, C.; Mascolo, A.; Gaio, M.; Sportiello, L.; Ferrajolo, C.; Rafaniello, C.; Rossi, F.; Capuano, A. PCSK9 Inhibitors and Neurocognitive Adverse Drug Reactions: Analysis of Individual Case Safety Reports from the Eudravigilance Database. Drug Saf. 2021, 44, 337–349. [Google Scholar] [CrossRef]
- Salaheldin, T.A.; Godugu, K.; Bharali, D.J.; Fujioka, K.; Elshourbagy, N.; Mousa, S.A. Novel oral nano-hepatic targeted anti-PCSK9 in hypercholesterolemia. Nanomedicine 2021, 40, 102480. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, J.; Jacome Sanz, D.; Ortutay, Z.; Seiskari, T.; Aittoniemi, J.; Huttunen, R.; Syrjanen, J.; Pesu, M. Reduced plasma PCSK9 response in patients with bacteraemia is associated with mortality. J. Intern. Med. 2019, 286, 553–561. [Google Scholar] [CrossRef]
- Pramfalk, C.; Jiang, Z.Y.; Parini, P. Hepatic Niemann-Pick C1-like 1. Curr. Opin. Lipidol. 2011, 22, 225–230. [Google Scholar] [CrossRef]
- Sudhop, T.; Reber, M.; Tribble, D.; Sapre, A.; Taggart, W.; Gibbons, P.; Musliner, T.; von Bergmann, K.; Lutjohann, D. Changes in cholesterol absorption and cholesterol synthesis caused by ezetimibe and/or simvastatin in men. J. Lipid Res. 2009, 50, 2117–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourimate, S.; Le May, C.; Langhi, C.; Jarnoux, A.L.; Ouguerram, K.; Zair, Y.; Nguyen, P.; Krempf, M.; Cariou, B.; Costet, P. Dual mechanisms for the fibrate-mediated repression of proprotein convertase subtilisin/kexin type 9. J. Biol. Chem. 2008, 283, 9666–9673. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.; Ancellin, N.; Charlton, F.; Comas, D.; Pilot, J.; Keech, A.; Patel, S.; Sullivan, D.R.; Cohn, J.S.; Rye, K.A.; et al. Plasma PCSK9 concentrations correlate with LDL and total cholesterol in diabetic patients and are decreased by fenofibrate treatment. Clin. Chem. 2008, 54, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Grewal, T.; Rentero, C.; Enrich, C.; Wahba, M.; Raabe, C.A.; Rescher, U. Annexin Animal Models-From Fundamental Principles to Translational Research. Int. J. Mol. Sci. 2021, 22, 3439. [Google Scholar] [CrossRef]
- Mayer, G.; Poirier, S.; Seidah, N.G. Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J. Biol. Chem. 2008, 283, 31791–31801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidah, N.G.; Poirier, S.; Denis, M.; Parker, R.; Miao, B.; Mapelli, C.; Prat, A.; Wassef, H.; Davignon, J.; Hajjar, K.A.; et al. Annexin A2 is a natural extrahepatic inhibitor of the PCSK9-induced LDL receptor degradation. PLoS ONE 2012, 7, e41865. [Google Scholar] [CrossRef] [Green Version]
- Fairoozy, R.H.; Cooper, J.; White, J.; Giambartolomei, C.; Folkersen, L.; Wannamethee, S.G.; Jefferis, B.J.; Whincup, P.; Ben-Shlomo, Y.; Kumari, M.; et al. Identifying low density lipoprotein cholesterol associated variants in the Annexin A2 (ANXA2) gene. Atherosclerosis 2017, 261, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Buechler, C.; Haberl, E.M.; Rein-Fischboeck, L.; Aslanidis, C. Adipokines in Liver Cirrhosis. Int. J. Mol. Sci. 2017, 18, 1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buechler, C.; Weiss, T.S. Does hepatic steatosis affect drug metabolizing enzymes in the liver? Curr. Drug Metab. 2011, 12, 24–34. [Google Scholar] [CrossRef]
- Tilg, H. The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Martin-Mateos, R.; Albillos, A. The Role of the Gut-Liver Axis in Metabolic Dysfunction-Associated Fatty Liver Disease. Front. Immunol. 2021, 12, 660179. [Google Scholar] [CrossRef]
- Rashid, S.; Tavori, H.; Brown, P.E.; Linton, M.F.; He, J.; Giunzioni, I.; Fazio, S. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation 2014, 130, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Garcon, D.; Moreau, F.; Ayer, A.; Dijk, W.; Prieur, X.; Arnaud, L.; Roubtsova, A.; Seidah, N.; Prat, A.; Cariou, B.; et al. Circulating Rather Than Intestinal PCSK9 (Proprotein Convertase Subtilisin Kexin Type 9) Regulates Postprandial Lipemia in Mice. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2084–2094. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Ben Djoudi Ouadda, A.; Spahis, S.; Sane, A.T.; Garofalo, C.; Grenier, E.; Emonnot, L.; Yara, S.; Couture, P.; Beaulieu, J.F.; et al. PCSK9 plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis 2013, 227, 297–306. [Google Scholar] [CrossRef]
- Peach, M.; Xu, R.; Fitzpatrick, D.; Hamilton, L.; Somaratne, R.; Scott, R.; Wasserman, S.M.; Djedjos, C.S. Effect of evolocumab on cholesterol synthesis and absorption. J. Lipid Res. 2016, 57, 2217–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C.; Watts, G.F.; Somaratne, R.; Wasserman, S.M.; Scott, R.; Barrett, P.H.R. Comparative Effects of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) Inhibition and Statins on Postprandial Triglyceride-Rich Lipoprotein Metabolism. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1644–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruscica, M.; Tokgozoglu, L.; Corsini, A.; Sirtori, C.R. PCSK9 inhibition and inflammation: A narrative review. Atherosclerosis 2019, 288, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Buechler, C.; Aslanidis, C. Role of lipids in pathophysiology, diagnosis and therapy of hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158658. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive Lipid Species and Metabolic Pathways in Progression and Resolution of Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 282–302.e8. [Google Scholar] [CrossRef]
- Ten Hove, M.; Pater, L.; Storm, G.; Weiskirchen, S.; Weiskirchen, R.; Lammers, T.; Bansal, R. The hepatic lipidome: From basic science to clinical translation. Adv. Drug Deliv. Rev. 2020, 159, 180–197. [Google Scholar] [CrossRef]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646, viii–ix. [Google Scholar] [CrossRef] [Green Version]
- De Ferranti, S.; Mozaffarian, D. The perfect storm: Obesity, adipocyte dysfunction, and metabolic consequences. Clin. Chem. 2008, 54, 945–955. [Google Scholar] [CrossRef] [Green Version]
- Tomkin, G.H.; Owens, D. Diabetes and dyslipidemia: Characterizing lipoprotein metabolism. Diabetes Metab. Syndr. Obes. 2017, 10, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Trinder, M.; Boyd, J.H.; Brunham, L.R. Molecular regulation of plasma lipid levels during systemic inflammation and sepsis. Curr. Opin. Lipidol. 2019, 30, 108–116. [Google Scholar] [CrossRef]
- Alborn, W.E.; Cao, G.; Careskey, H.E.; Qian, Y.W.; Subramaniam, D.R.; Davies, J.; Conner, E.M.; Konrad, R.J. Serum proprotein convertase subtilisin kexin type 9 is correlated directly with serum LDL cholesterol. Clin. Chem. 2007, 53, 1814–1819. [Google Scholar] [CrossRef] [Green Version]
- Nozue, T.; Hattori, H.; Ogawa, K.; Kujiraoka, T.; Iwasaki, T.; Hirano, T.; Michishita, I. Correlation between serum levels of proprotein convertase subtilisin/kexin type 9 (PCSK9) and atherogenic lipoproteins in patients with coronary artery disease. Lipids Health Dis. 2016, 15, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardiola, M.; Plana, N.; Ibarretxe, D.; Cabre, A.; Gonzalez, M.; Ribalta, J.; Masana, L. Circulating PCSK9 levels are positively correlated with NMR-assessed atherogenic dyslipidaemia in patients with high cardiovascular risk. Clin. Sci. 2015, 128, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Ferri, N.; Macchi, C.; Meroni, M.; Lanti, C.; Ricci, C.; Maggioni, M.; Fracanzani, A.L.; Badiali, S.; Fargion, S.; et al. Liver fat accumulation is associated with circulating PCSK9. Ann. Med. 2016, 48, 384–391. [Google Scholar] [CrossRef]
- Gu, Q.; Paulose-Ram, R.; Burt, V.L.; Kit, B.K. Prescription cholesterol-lowering medication use in adults aged 40 and over: United States, 2003–2012. NCHS Data Brief. 2014, 177, 1–8. [Google Scholar]
- Knopf, H.C.; Busch, M.A.; Du, Y.; Truthmann, J.; Schienkiewitz, A.; Scheidt-Nave, C. Changes in the prevalence of statin use in Germany—Findings from national health interview and examination surveys 1997–1999 and 2008–2011. Z. Evidenz Fortbild. Qual. Gesundh. 2017, 122, 22–31. [Google Scholar] [CrossRef]
- Bassani, L.; Fernandes, S.A.; Raimundo, F.V.; Harter, D.L.; Gonzalez, M.C.; Marroni, C.A. Lipid Profile of Cirrhotic Patients and Its Association with Prognostic Scores: A Cross-Sectional Study. Arq. Gastroenterol. 2015, 52, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Adinolfi, L.E.; Loria, P.; Carulli, N.; Ruggiero, G.; Day, C.P. Steatosis and hepatitis C virus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology 2004, 126, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Benn, M.; Nordestgaard, B.G.; Grande, P.; Schnohr, P.; Tybjaerg-Hansen, A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J. Am. Coll. Cardiol. 2010, 55, 2833–2842. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef]
- Janis, M.T.; Tarasov, K.; Ta, H.X.; Suoniemi, M.; Ekroos, K.; Hurme, R.; Lehtimaki, T.; Paiva, H.; Kleber, M.E.; Marz, W.; et al. Beyond LDL-C lowering: Distinct molecular sphingolipids are good indicators of proprotein convertase subtilisin/kexin type 9 (PCSK9) deficiency. Atherosclerosis 2013, 228, 380–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammad, S.M.; Pierce, J.S.; Soodavar, F.; Smith, K.J.; Al Gadban, M.M.; Rembiesa, B.; Klein, R.L.; Hannun, Y.A.; Bielawski, J.; Bielawska, A. Blood sphingolipidomics in healthy humans: Impact of sample collection methodology. J. Lipid Res. 2010, 51, 3074–3087. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, P.; Leidl, K.; Boettcher, A.; Schmitz, G.; Liebisch, G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J. Lipid Res. 2009, 50, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Hilvo, M.; Simolin, H.; Metso, J.; Ruuth, M.; Oorni, K.; Jauhiainen, M.; Laaksonen, R.; Baruch, A. PCSK9 inhibition alters the lipidome of plasma and lipoprotein fractions. Atherosclerosis 2018, 269, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Agarwala, A.P.; Rodrigues, A.; Risman, M.; McCoy, M.; Trindade, K.; Qu, L.; Cuchel, M.; Billheimer, J.; Rader, D.J. High-Density Lipoprotein (HDL) Phospholipid Content and Cholesterol Efflux Capacity Are Reduced in Patients With Very High HDL Cholesterol and Coronary Disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1515–1519. [Google Scholar] [CrossRef] [Green Version]
- Kamada, Y.; Takehara, T.; Hayashi, N. Adipocytokines and liver disease. J. Gastroenterol. 2008, 43, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Wanninger, J.; Neumeier, M. Adiponectin, a key adipokine in obesity related liver diseases. World J. Gastroenterol. 2011, 17, 2801–2811. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Li, S.; Cui, C.J.; Zhang, Y.; Yang, S.H.; Li, J.J. Leptin decreases the expression of low-density lipoprotein receptor via PCSK9 pathway: Linking dyslipidemia with obesity. J. Transl. Med. 2016, 14, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchi, C.; Greco, M.F.; Botta, M.; Sperandeo, P.; Dongiovanni, P.; Valenti, L.; Cicero AF, G.; Borghi, C.; Lupo, M.G.; Romeo, S.; et al. Leptin, Resistin, and Proprotein Convertase Subtilisin/Kexin Type 9: The Role of STAT3. Am. J. Pathol. 2020, 190, 2226–2236. [Google Scholar] [CrossRef]
- Dessie, G.; Ayelign, B.; Akalu, Y.; Shibabaw, T.; Molla, M.D. Effect of Leptin on Chronic Inflammatory Disorders: Insights to Therapeutic Target to Prevent Further Cardiovascular Complication. Diabetes Metab. Syndr. Obes. 2021, 14, 3307–3322. [Google Scholar] [CrossRef]
- Ruscica, M.; Ricci, C.; Macchi, C.; Magni, P.; Cristofani, R.; Liu, J.; Corsini, A.; Ferri, N. Suppressor of Cytokine Signaling-3 (SOCS-3) Induces Proprotein Convertase Subtilisin Kexin Type 9 (PCSK9) Expression in Hepatic HepG2 Cell Line. J. Biol. Chem. 2016, 291, 3508–3519. [Google Scholar] [CrossRef] [Green Version]
- Cao, A.; Wu, M.; Li, H.; Liu, J. Janus kinase activation by cytokine oncostatin M decreases PCSK9 expression in liver cells. J. Lipid Res. 2011, 52, 518–530. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; Tsurusaki, S.; Miyata, N.; Saijou, E.; Okochi, H.; Miyajima, A.; Tanaka, M. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology 2018, 67, 296–312. [Google Scholar] [CrossRef] [Green Version]
- Thorlacius-Ussing, G.; Nielsen, B.; Tougaard, P.; Andersen, E.; Olsen, J.; Pedersen, A. Interleukin-1β Regulates PCSK9 and LDL Receptor Expression together with de novo Cholesterol Synthesis in HepG2 Cells. Glob. J. Gastroenterol. Hepatol. 2016, 4, 36–44. [Google Scholar]
- Ueland, T.; Kleveland, O.; Michelsen, A.E.; Wiseth, R.; Damas, J.K.; Aukrust, P.; Gullestad, L.; Halvorsen, B.; Yndestad, A. Serum PCSK9 is modified by interleukin-6 receptor antagonism in patients with hypercholesterolaemia following non-ST-elevation myocardial infarction. Open Heart 2018, 5, e000765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Yang, X.; Li, Q.; Zeng, P.; Liu, Y.; Liu, L.; Chen, Y.; Yu, M.; Ma, C.; Li, X.; et al. Activation of Adiponectin Receptor Regulates Proprotein Convertase Subtilisin/Kexin Type 9 Expression and Inhibits Lesions in ApoE-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, K.R.; Moser, A.H.; Shigenaga, J.K.; Patzek, S.M.; Grunfeld, C. Inflammation stimulates the expression of PCSK9. Biochem. Biophys. Res. Commun. 2008, 374, 341–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momtazi-Borojeni, A.A.; Sabouri-Rad, S.; Gotto, A.M.; Pirro, M.; Banach, M.; Awan, Z.; Barreto, G.E.; Sahebkar, A. PCSK9 and inflammation: A review of experimental and clinical evidence. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 237–245. [Google Scholar] [CrossRef]
- Walley, K.R.; Thain, K.R.; Russell, J.A.; Reilly, M.P.; Meyer, N.J.; Ferguson, J.F.; Christie, J.D.; Nakada, T.A.; Fjell, C.D.; Thair, S.A.; et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci. Transl. Med. 2014, 6, 258ra143. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Li, X.; Yuan, Y.J.; Chen, Z.L.; He, J.H.; Wu, J.H.; Cai, X.S. Inhibition of proprotein convertase subtilisin/kexin type 9 attenuates 2,4,6-trinitrobenzenesulfonic acid-induced colitis via repressing toll-like receptor 4/nuclear factor-kappa B. Kaohsiung J. Med. Sci. 2020, 36, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, D.J.; Grin, P.M.; Khan, M.; Prat, A.; Zhou, J.; Fox-Robichaud, A.E.; Seidah, N.G.; Liaw, P.C. Differential Expression of PCSK9 Modulates Infection, Inflammation, and Coagulation in a Murine Model of Sepsis. Shock 2016, 46, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.H.; Fjell, C.D.; Russell, J.A.; Sirounis, D.; Cirstea, M.S.; Walley, K.R. Increased Plasma PCSK9 Levels Are Associated with Reduced Endotoxin Clearance and the Development of Acute Organ Failures during Sepsis. J. Innate. Immun. 2016, 8, 211–220. [Google Scholar] [CrossRef]
- Mitchell, K.A.; Moore, J.X.; Rosenson, R.S.; Irvin, R.; Guirgis, F.W.; Shapiro, N.; Safford, M.; Wang, H.E. PCSK9 loss-of-function variants and risk of infection and sepsis in the Reasons for Geographic and Racial Differences in Stroke (REGARDS) cohort. PLoS ONE 2019, 14, e0210808. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, X.J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef]
- Holt, A.P.; Salmon, M.; Buckley, C.D.; Adams, D.H. Immune interactions in hepatic fibrosis. Clin. Liver Dis. 2008, 12, 861–882. [Google Scholar] [CrossRef] [Green Version]
- Ricci, C.; Ruscica, M.; Camera, M.; Rossetti, L.; Macchi, C.; Colciago, A.; Zanotti, I.; Lupo, M.G.; Adorni, M.P.; Cicero, A.F.G.; et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci. Rep. 2018, 8, 2267. [Google Scholar] [CrossRef] [Green Version]
- Jacome Sanz, D.; Saralahti, A.K.; Pekkarinen, M.; Kesseli, J.; Nykter, M.; Ramet, M.; Ojanen MJ, T.; Pesu, M. Proprotein convertase subtilisin/kexin type 9 regulates the production of acute-phase reactants from the liver. Liver Int. 2021, 41, 2511–2522. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Massoud, O.; Charlton, M. Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Clin. Liver Dis. 2018, 22, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.E.; Serper, M.A.; Mehta, R.; Fox, R.; John, B.; Aytaman, A.; Baytarian, M.; Hunt, K.; Albrecht, J.; Njei, B.; et al. Effects of Hypercholesterolemia and Statin Exposure on Survival in a Large National Cohort of Patients With Cirrhosis. Gastroenterology 2019, 156, 1693–1706.e12. [Google Scholar] [CrossRef] [Green Version]
- Clugston, R.D.; Gao, M.A.; Blaner, W.S. The Hepatic Lipidome: A Gateway to Understanding the Pathogenes is of Alcohol-Induced Fatty Liver. Curr. Mol. Pharmacol. 2017, 10, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Orman, E.S.; Odena, G.; Bataller, R. Alcoholic liver disease: Pathogenesis, management, and novel targets for therapy. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 77–84. [Google Scholar] [CrossRef] [Green Version]
- Day, C.P.; James, O.F. Hepatic steatosis: Innocent bystander or guilty party? Hepatology 1998, 27, 1463–1466. [Google Scholar] [CrossRef]
- Visioli, F.; Monti, S.; Colombo, C.; Galli, C. Ethanol enhances cholesterol synthesis and secretion in human hepatomal cells. Alcohol 1998, 15, 299–303. [Google Scholar] [CrossRef]
- Zeng, T.; Zhang, C.L.; Xiao, M.; Yang, R.; Xie, K.Q. Critical Roles of Kupffer Cells in the Pathogenesis of Alcoholic Liver Disease: From Basic Science to Clinical Trials. Front. Immunol. 2016, 7, 538. [Google Scholar] [CrossRef]
- Louvet, A.; Teixeira-Clerc, F.; Chobert, M.N.; Deveaux, V.; Pavoine, C.; Zimmer, A.; Pecker, F.; Mallat, A.; Lotersztajn, S. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 2011, 54, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Lohoff, F.W.; Sorcher, J.L.; Rosen, A.D.; Mauro, K.L.; Fanelli, R.R.; Momenan, R.; Hodgkinson, C.A.; Vendruscolo, L.F.; Koob, G.F.; Schwandt, M.; et al. Methylomic profiling and replication implicates deregulation of PCSK9 in alcohol use disorder. Mol. Psychiatry 2018, 23, 1900–1910. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, M.; Angeli, P.; Claria, J.; Moreau, R.; Gines, P.; Jalan, R.; Caraceni, P.; Fernandez, J.; Gerbes, A.L.; O’Brien, A.J.; et al. Albumin in decompensated cirrhosis: New concepts and perspectives. Gut 2020, 69, 1127–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Mukhopadhyay, P.; Matyas, C.; Trojnar, E.; Paloczi, J.; Yang, Y.R.; Blank, B.A.; Savage, C.; Sorokin, A.V.; Mehta, N.N.; et al. PCSK9 inhibition as a novel therapeutic target for alcoholic liver disease. Sci. Rep. 2019, 9, 17167. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Huang, M.H.; Jiang, J.D.; Peng, Z.G. Hepatitis C: From inflammatory pathogenesis to anti-inflammatory/hepatoprotective therapy. World J. Gastroenterol. 2018, 24, 5297–5311. [Google Scholar] [CrossRef]
- Sarrazin, C. Treatment failure with DAA therapy: Importance of resistance. J. Hepatol. 2021, 74, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Di Caprio, G.; Fimia, G.M.; Ippolito, G.; Tripodi, M.; Alonzi, T. Hepatitis C virus relies on lipoproteins for its life cycle. World J. Gastroenterol. 2016, 22, 1953–1965. [Google Scholar] [CrossRef]
- Labonte, P.; Begley, S.; Guevin, C.; Asselin, M.C.; Nassoury, N.; Mayer, G.; Prat, A.; Seidah, N.G. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology 2009, 50, 17–24. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Q. Proprotein convertase subtilisin/kexin type 9 inhibits hepatitis C virus replication through interacting with NS5A. J. Gen. Virol. 2018, 99, 44–61. [Google Scholar] [CrossRef]
- Hyrina, A.; Olmstead, A.D.; Steven, P.; Krajden, M.; Tam, E.; Jean, F. Treatment-Induced Viral Cure of Hepatitis C Virus-Infected Patients Involves a Dynamic Interplay among three Important Molecular Players in Lipid Homeostasis: Circulating microRNA (miR)-24, miR-223, and Proprotein Convertase Subtilisin/Kexin Type 9. EBioMedicine 2017, 23, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, A.; Gusarova, V.; Stahl, N.; Gurnett-Bander, A.; Kyratsous, C.A. Alirocumab, a Therapeutic Human Antibody to PCSK9, Does Not Affect CD81 Levels or Hepatitis C Virus Entry and Replication into Hepatocytes. PLoS ONE 2016, 11, e0154498. [Google Scholar] [CrossRef] [Green Version]
- Zapatero-Belinchon, F.J.; Otjengerdes, R.; Sheldon, J.; Schulte, B.; Carriqui-Madronal, B.; Brogden, G.; Arroyo-Fernandez, L.M.; Vondran FW, R.; Maasoumy, B.; von Hahn, T.; et al. Interdependent Impact of Lipoprotein Receptors and Lipid-Lowering Drugs on HCV Infectivity. Cells 2021, 10, 1626. [Google Scholar] [CrossRef] [PubMed]
- Syed, G.H.; Tang, H.; Khan, M.; Hassanein, T.; Liu, J.; Siddiqui, A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J. Virol. 2014, 88, 2519–2529. [Google Scholar] [CrossRef] [Green Version]
- Fasolato, S.; Pigozzo, S.; Pontisso, P.; Angeli, P.; Ruscica, M.; Savarino, E.; De Martin, S.; Lupo, M.G.; Ferri, N. PCSK9 Levels Are Raised in Chronic HCV Patients with Hepatocellular Carcinoma. J. Clin. Med. 2020, 9, 3134. [Google Scholar] [CrossRef] [PubMed]
- Torti, C.; Scaglione, V.; Cesana, B.M.; Costa, C.; Marascio, N.; Schiaroli, E.; Busti, C.; Bastianelli, S.; Mazzitelli, M.; Trecarichi, E.M.; et al. Effect of directly acting antivirals for hepatitis C virus infection on proprotein convertase subtilisin/kexin type 9 level. Health Sci. Rep. 2021, 4, e273. [Google Scholar] [CrossRef]
- Bridge, S.H.; Sheridan, D.A.; Felmlee, D.J.; Crossey, M.M.; Fenwick, F.I.; Lanyon, C.V.; Dubuc, G.; Seidah, N.G.; Davignon, J.; Thomas, H.C.; et al. PCSK9, apolipoprotein E and lipoviral particles in chronic hepatitis C genotype 3: Evidence for genotype-specific regulation of lipoprotein metabolism. J. Hepatol. 2015, 62, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Miyaaki, H.; Miuma, S.; Taura, N.; Motoyoshi, Y.; Akahoshi, H.; Nakamura, J.; Takahashi, Y.; Honda, T.; Yajima, H.; et al. Changes in serum LDL, PCSK9 and microRNA-122 in patients with chronic HCV infection receiving Daclatasvir/Asunaprevir. Biomed. Rep. 2019, 10, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, J.; Peschel, G.; Müller, M.; Schacherer, D.; Wiest, R.; Weigand, K.; Buechler, C. Rapid Decline of Serum Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) in Non-Cirrhotic Patients with Chronic Hepatitis C Infection Receiving Direct-Acting Antiviral Therapy. J. Clin. Med. 2021, 10, 1621. [Google Scholar] [CrossRef]
- Kohli, P.; Ganz, P.; Ma, Y.; Scherzer, R.; Hur, S.; Weigel, B.; Grunfeld, C.; Deeks, S.; Wasserman, S.; Scott, R.; et al. HIV and Hepatitis C-Coinfected Patients Have Lower Low-Density Lipoprotein Cholesterol Despite Higher Proprotein Convertase Subtilisin Kexin 9 (PCSK9): An Apparent “PCSK9-Lipid Paradox”. J. Am. Heart Assoc. 2016, 5, e002683. [Google Scholar] [CrossRef] [Green Version]
- Graf, C.; Welzel, T.; Bogdanou, D.; Vermehren, J.; Beckel, A.; Bojunga, J.; Friedrich-Rust, M.; Dietz, J.; Kubesch, A.; Mondorf, A.; et al. Hepatitis C Clearance by Direct-Acting Antivirals Impacts Glucose and Lipid Homeostasis. J. Clin. Med. 2020, 9, 2702. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Yatsuhashi, H.; Abiru, S.; Yamasaki, K.; Komori, A.; Nagaoka, S.; Saeki, A.; Uchida, S.; Bekki, S.; Kugiyama, Y.; et al. Rapid Increase in Serum Low-Density Lipoprotein Cholesterol Concentration during Hepatitis C Interferon-Free Treatment. PLoS ONE 2016, 11, e0163644. [Google Scholar] [CrossRef]
- Iossa, D.; Vitrone, M.; Gagliardi, M.; Falco, E.; Ragone, E.; Zampino, R.; Durante-Mangoni, E. Anthropometric parameters and liver histology influence lipid metabolic changes in HCV chronic hepatitis on direct-acting antiviral treatment. Ann. Transl. Med. 2021, 9, 35. [Google Scholar] [CrossRef]
- Pedersen, M.R.; Patel, A.; Backstedt, D.; Choi, M.; Seetharam, A.B. Genotype specific peripheral lipid profile changes with hepatitis C therapy. World J. Gastroenterol. 2016, 22, 10226–10231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Soroida, Y.; Sato, M.; Hikita, H.; Kobayashi, T.; Endo, M.; Sato, M.; Gotoh, H.; Iwai, T.; Tateishi, R.; et al. Eradication of hepatitis C virus is associated with the attenuation of steatosis as evaluated using a controlled attenuation parameter. Sci. Rep. 2018, 8, 7845. [Google Scholar] [CrossRef]
- Peschel, G.; Grimm, J.; Gulow, K.; Muller, M.; Buechler, C.; Weigand, K. Chemerin Is a Valuable Biomarker in Patients with HCV Infection and Correlates with Liver Injury. Diagnostics 2020, 10, 974. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, P.F.; Byun, J.H.; Platko, K.; Al-Hashimi, A.A.; Lhotak, S.; MacDonald, M.E.; Mejia-Benitez, A.; Prat, A.; Igdoura, S.A.; Trigatti, B.; et al. Pcsk9 knockout exacerbates diet-induced non-alcoholic steatohepatitis, fibrosis and liver injury in mice. JHEP Rep. 2019, 1, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Alem, S.A.; Abdellatif, Z.; Mabrouk, M.; Zayed, N.; Elsharkawy, A.; Khairy, M.; Musa, S.; Anwar, I.; Yosry, A. Diagnostic accuracy of acoustic radiation force impulse elastography (ARFI) in comparison to other non-invasive modalities in staging of liver fibrosis in chronic HCV patients: Single-center experience. Abdom. Radiol. (N. Y.) 2019, 44, 2751–2758. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. Electronic address e. e. e. EASL-ALEH Clinical Practice Guidelines: Non-invasive testsfor evaluation of liver disease severity and prognosis. J. Hepatol. 2015, 63, 237–265. [Google Scholar] [CrossRef] [Green Version]
- Paranagua-Vezozzo, D.C.; Andrade, A.; Mazo, D.F.; Nunes, V.; Guedes, A.L.; Ragazzo, T.G.; Moutinho, R.; Nacif, L.S.; Ono, S.K.; Alves, V.A.; et al. Concordance of non-invasive mechanical and serum tests for liver fibrosis evaluation in chronic hepatitis C. World J. Hepatol. 2017, 9, 436–442. [Google Scholar] [CrossRef]
- Carmona, I.; Cordero, P.; Ampuero, J.; Rojas, A.; Romero-Gomez, M. Role of assessing liver fibrosis in management of chronic hepatitis C virus infection. Clin. Microbiol. Infect. 2016, 22, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Yeh, M.M.; Brunt, E.M. Pathological features of fatty liver disease. Gastroenterology 2014, 147, 754–764. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- He, Y.; Rodrigues, R.M.; Wang, X.; Seo, W.; Ma, J.; Hwang, S.; Fu, Y.; Trojnar, E.; Matyas, C.; Zhao, S.; et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J. Clin. Investig. 2021, 131, e141513. [Google Scholar] [CrossRef]
- Jha, P.; Knopf, A.; Koefeler, H.; Mueller, M.; Lackner, C.; Hoefler, G.; Claudel, T.; Trauner, M. Role of adipose tissue in methionine-choline-deficient model of non-alcoholic steatohepatitis (NASH). Biochim. Biophys. Acta 2014, 1842, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Takahashi, S.; Fang, Z.Z.; Matsubara, T.; Krausz, K.W.; Qu, A.; Gonzalez, F.J. Role of white adipose lipolysis in the development of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta 2014, 1841, 1596–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattenberg, J.M.; Galle, P.R. Animal models of non-alcoholic steatohepatitis: Of mice and man. Dig. Dis. 2010, 28, 247–254. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Lee, S.P.; Linsley, P.S.; Gersuk, V.; Yeh, M.M.; Chen, Y.Y.; Peng, Y.J.; Dutta, M.; Mascarinas, G.; Molla, B.; et al. Pcsk9 Deletion Promotes Murine Nonalcoholic Steatohepatitis and Hepatic Carcinogenesis: Role of Cholesterol. Hepatol. Commun. 2021. [Google Scholar] [CrossRef] [PubMed]
- Baragetti, A.; Balzarotti, G.; Grigore, L.; Pellegatta, F.; Guerrini, U.; Pisano, G.; Fracanzani, A.L.; Fargion, S.; Norata, G.D.; Catapano, A.L. PCSK9 deficiency results in increased ectopic fat accumulation in experimental models and in humans. Eur. J. Prev. Cardiol. 2017, 24, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Roubtsova, A.; Munkonda, M.N.; Awan, Z.; Marcinkiewicz, J.; Chamberland, A.; Lazure, C.; Cianflone, K.; Seidah, N.G.; Prat, A. Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, A.; Samami, S.; Lauzier, B.; Des Rosiers, C.; Ngo Sock, E.T.; Ong, H.; Mayer, G. PCSK9 Induces CD36 Degradation and Affects Long-Chain Fatty Acid Uptake and Triglyceride Metabolism in Adipocytes and in Mouse Liver. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2517–2525. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Gonzalez-Rodriguez, A.; Garcia-Monzon, C.; Valverde, A.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, M.; Walmann, T.; Nutalapati, V.; Gibson, C.; Zafar, Y. Effects of proprotein convertase subtilisin/kexin type-9 inhibitors on fatty liver. World J. Hepatol. 2020, 12, 1258–1266. [Google Scholar] [CrossRef]
- Dimakopoulou, A.; Sfikas, G.; Athyros, V. PCSK9 administration ameliorates nonalcoholic fatty disease in patients with heterozygous familial hyperlipidemia. Hell. J. Atheroscler. 2018, 9. [Google Scholar] [CrossRef]
- Scicali, R.; Di Pino, A.; Urbano, F.; Ferrara, V.; Marchisello, S.; Di Mauro, S.; Scamporrino, A.; Filippello, A.; Rabuazzo, A.M.; Purrello, F.; et al. Analysis of steatosis biomarkers and inflammatory profile after adding on PCSK9 inhibitor treatment in familial hypercholesterolemia subjects with nonalcoholic fatty liver disease: A single lipid center real-world experience. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Kotowski, I.K.; Pertsemlidis, A.; Luke, A.; Cooper, R.S.; Vega, G.L.; Cohen, J.C.; Hobbs, H.H. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 2006, 78, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Lebeau, P.F.; Wassef, H.; Byun, J.H.; Platko, K.; Ason, B.; Jackson, S.; Dobroff, J.; Shetterly, S.; Richards, W.G.; Al-Hashimi, A.A.; et al. The loss-of-function PCSK9Q152H variant increases ER chaperones GRP78 and GRP94 and protects against liver injury. J. Clin. Investig. 2021, 131, e128650. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Gawrieh, S.; Liang, T.; McIntyre, A.D.; Hegele, R.A.; Chalasani, N. Interrogation of selected genes influencing serum LDL-Cholesterol levels in patients with well characterized NAFLD. J. Clin. Lipidol. 2021, 15, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Wargny, M.; Ducluzeau, P.H.; Petit, J.M.; Le May, C.; Smati, S.; Arnaud, L.; Pichelin, M.; Bouillet, B.; Lannes, A.; Blanchet, O.; et al. Circulating PCSK9 levels are not associated with the severity of hepatic steatosis and NASH in a high-risk population. Atherosclerosis 2018, 278, 82–90. [Google Scholar] [CrossRef]
- Emma, M.R.; Giannitrapani, L.; Cabibi, D.; Porcasi, R.; Pantuso, G.; Augello, G.; Giglio, R.V.; Re, N.L.; Capitano, A.R.; Montalto, G.; et al. Hepatic and circulating levels of PCSK9 in morbidly obese patients: Relation with severity of liver steatosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158792. [Google Scholar] [CrossRef]
- Paquette, M.; Gauthier, D.; Chamberland, A.; Prat, A.; De Lucia Rolfe, E.; Rasmussen, J.J.; Kaduka, L.; Seidah, N.G.; Bernard, S.; Christensen, D.L.; et al. Circulating PCSK9 is associated with liver biomarkers and hepatic steatosis. Clin. Biochem. 2020, 77, 20–25. [Google Scholar] [CrossRef]
- Geier, A. Hepatitis B virus: The “metabolovirus” highjacks cholesterol and bile acid metabolism. Hepatology 2014, 60, 1458–1460. [Google Scholar] [CrossRef]
- Li, Y.; Luo, G. Human low-density lipoprotein receptor plays an important role in hepatitis B virus infection. PLoS Pathog. 2021, 17, e1009722. [Google Scholar] [CrossRef]
- Nagashima, S.; Morishima, K.; Okamoto, H.; Ishibashi, S. Possible involvement of PCSK9 overproduction in hyperlipoproteinemia associated with hepatocellular carcinoma: A case report. J. Clin. Lipidol. 2016, 10, 1045–1049. [Google Scholar] [CrossRef]
- Beyoglu, D.; Idle, J.R. The metabolomic window into hepatobiliary disease. J. Hepatol. 2013, 59, 842–858. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Z.; Zhu, X.D.; Feng, L.H.; Li, X.L.; Liu, X.F.; Sun, H.C.; Tang, Z.Y. PCSK9 promotes tumor growth by inhibiting tumor cell apoptosis in hepatocellular carcinoma. Exp. Hematol. Oncol. 2021, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Pope, E.D., 3rd; Kimbrough, E.O.; Vemireddy, L.P.; Surapaneni, P.K.; Copland, J.A., 3rd; Mody, K. Aberrant lipid metabolism as a therapeutic target in liver cancer. Expert Opin. Ther. Targets 2019, 23, 473–483. [Google Scholar] [CrossRef]
- Sun, X.; Essalmani, R.; Day, R.; Khatib, A.M.; Seidah, N.G.; Prat, A. Proprotein convertase subtilisin/kexin type 9 deficiency reduces melanoma metastasis in liver. Neoplasia 2012, 14, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Hu, J.; Fang, T.; Tang, W.; Lv, B.; Yang, B.; Xia, J. Protein convertase subtilisin/Kexin type 9 inhibits hepatocellular carcinoma growth by interacting with GSTP1 and suppressing the JNK signaling pathway. Cancer Biol. Med. 2021, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Proteinatlas The Human Protein Atlas. Available online: https://www.proteinatlas.org (accessed on 19 December 2019).
- Mahboobnia, K.; Pirro, M.; Marini, E.; Grignani, F.; Bezsonov, E.E.; Jamialahmadi, T.; Sahebkar, A. PCSK9 and cancer: Rethinking the link. Biomed. Pharmacother. 2021, 140, 111758. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Hoque, M.; Rentero, C.; Conway, J.R.; Murray, R.Z.; Timpson, P.; Enrich, C.; Grewal, T. The cross-talk of LDL-cholesterol with cell motility: Insights from the Niemann Pick Type C1 mutation and altered integrin trafficking. Cell Adh. Migr. 2015, 9, 384–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angel, A.; D’Costa, M.A.; Yuen, R. Low density lipoprotein binding, internalization, and degradation in human adipose cells. Can. J. Biochem. 1979, 57, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, S.; Ferre, P.; Dugail, I. Adipocyte cholesterol balance in obesity. Biochem. Soc. Trans. 2004, 32, 103–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraj, M. LDL, LDL receptors, and PCSK9 as modulators of the risk for type 2 diabetes: A focus on white adipose tissue. J. Biomed. Res. 2020, 34, 251–259. [Google Scholar] [CrossRef]
- Bordicchia, M.; Spannella, F.; Ferretti, G.; Bacchetti, T.; Vignini, A.; Di Pentima, C.; Mazzanti, L.; Sarzani, R. PCSK9 is Expressed in Human Visceral Adipose Tissue and Regulated by Insulin and Cardiac Natriuretic Peptides. Int. J. Mol. Sci. 2019, 20, 245. [Google Scholar] [CrossRef] [Green Version]
- Coburn, C.T.; Knapp, F.F., Jr.; Febbraio, M.; Beets, A.L.; Silverstein, R.L.; Abumrad, N.A. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 2000, 275, 32523–32529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmas, C.E.; Silverio, D.; Sourlas, A.; Garcia, F.; Montan, P.D.; Guzman, E. Impact of lipid-lowering therapy on glycemic control and the risk for new-onset diabetes mellitus. Drugs Context 2018, 7, 212562. [Google Scholar] [CrossRef] [Green Version]
- Klimentidis, Y.C.; Arora, A.; Newell, M.; Zhou, J.; Ordovas, J.M.; Renquist, B.J.; Wood, A.C. Phenotypic and Genetic Characterization of Lower LDL Cholesterol and Increased Type 2 Diabetes Risk in the UK Biobank. Diabetes 2020, 69, 2194–2205. [Google Scholar] [CrossRef]
- Cyr, Y.; Lamantia, V.; Bissonnette, S.; Burnette, M.; Besse-Patin, A.; Demers, A.; Wabitsch, M.; Chretien, M.; Mayer, G.; Estall, J.L.; et al. Lower plasma PCSK9 in normocholesterolemic subjects is associated with upregulated adipose tissue surface-expression of LDLR and CD36 and NLRP3 inflammasome. Physiol. Rep. 2021, 9, e14721. [Google Scholar] [CrossRef]
- Krautbauer, S.; Neumeier, M.; Eisinger, K.; Hader, Y.; Dada, A.; Schmitz, G.; Aslanidis, C.; Buechler, C. LDL but not HDL increases adiponectin release of primary human adipocytes. Exp. Mol. Pathol. 2013, 95, 325–329. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grewal, T.; Buechler, C. Emerging Insights on the Diverse Roles of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Chronic Liver Diseases: Cholesterol Metabolism and Beyond. Int. J. Mol. Sci. 2022, 23, 1070. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031070
Grewal T, Buechler C. Emerging Insights on the Diverse Roles of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Chronic Liver Diseases: Cholesterol Metabolism and Beyond. International Journal of Molecular Sciences. 2022; 23(3):1070. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031070
Chicago/Turabian StyleGrewal, Thomas, and Christa Buechler. 2022. "Emerging Insights on the Diverse Roles of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Chronic Liver Diseases: Cholesterol Metabolism and Beyond" International Journal of Molecular Sciences 23, no. 3: 1070. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031070