
In Silico and Experimental ADAM17 Kinetic Modeling as Basis for Future Screening System for Modulators

Abstract
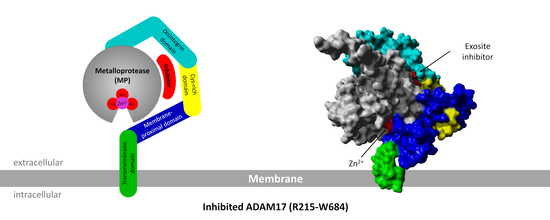
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Experimental Pharmacology
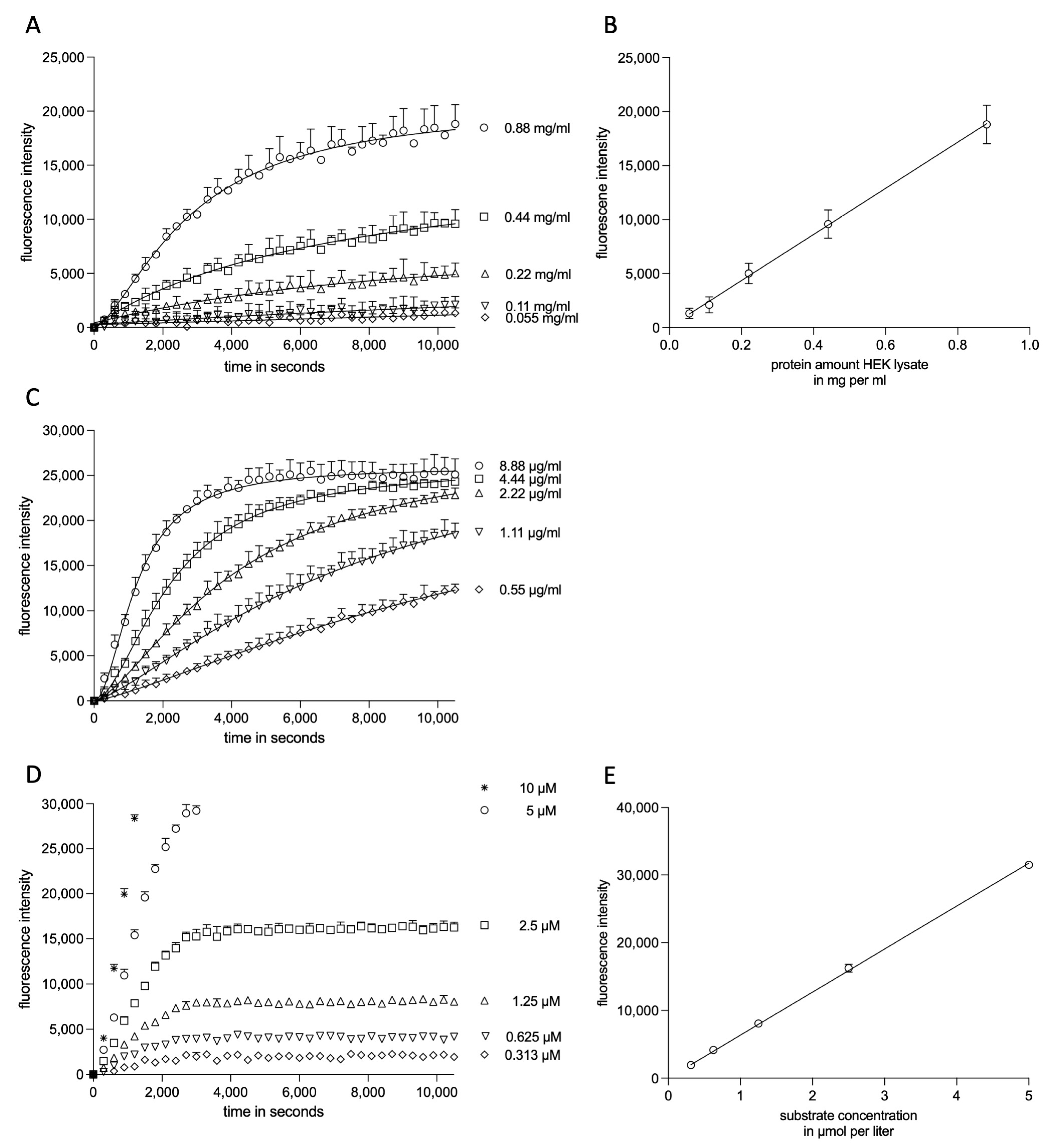
2.1.1. Experimental Kinetic Modeling Setup
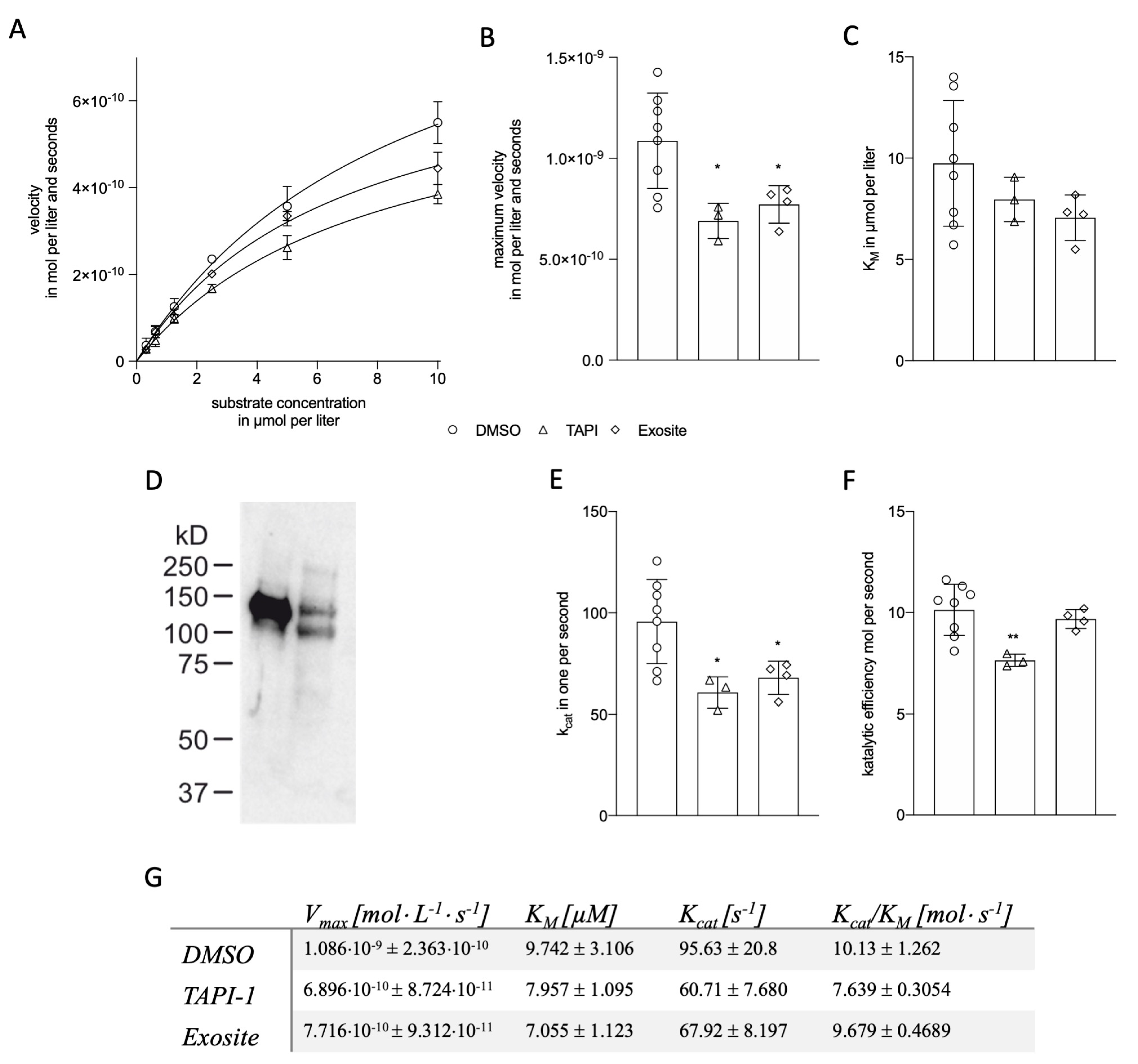
2.1.2. Kinetic Modeling of ADAM17 Inhibitors
2.1.3. Kinetic Modelling as Predictive Tool for ADAM17 Modulators
2.2. 3D Structure Modeling and Molecular Docking
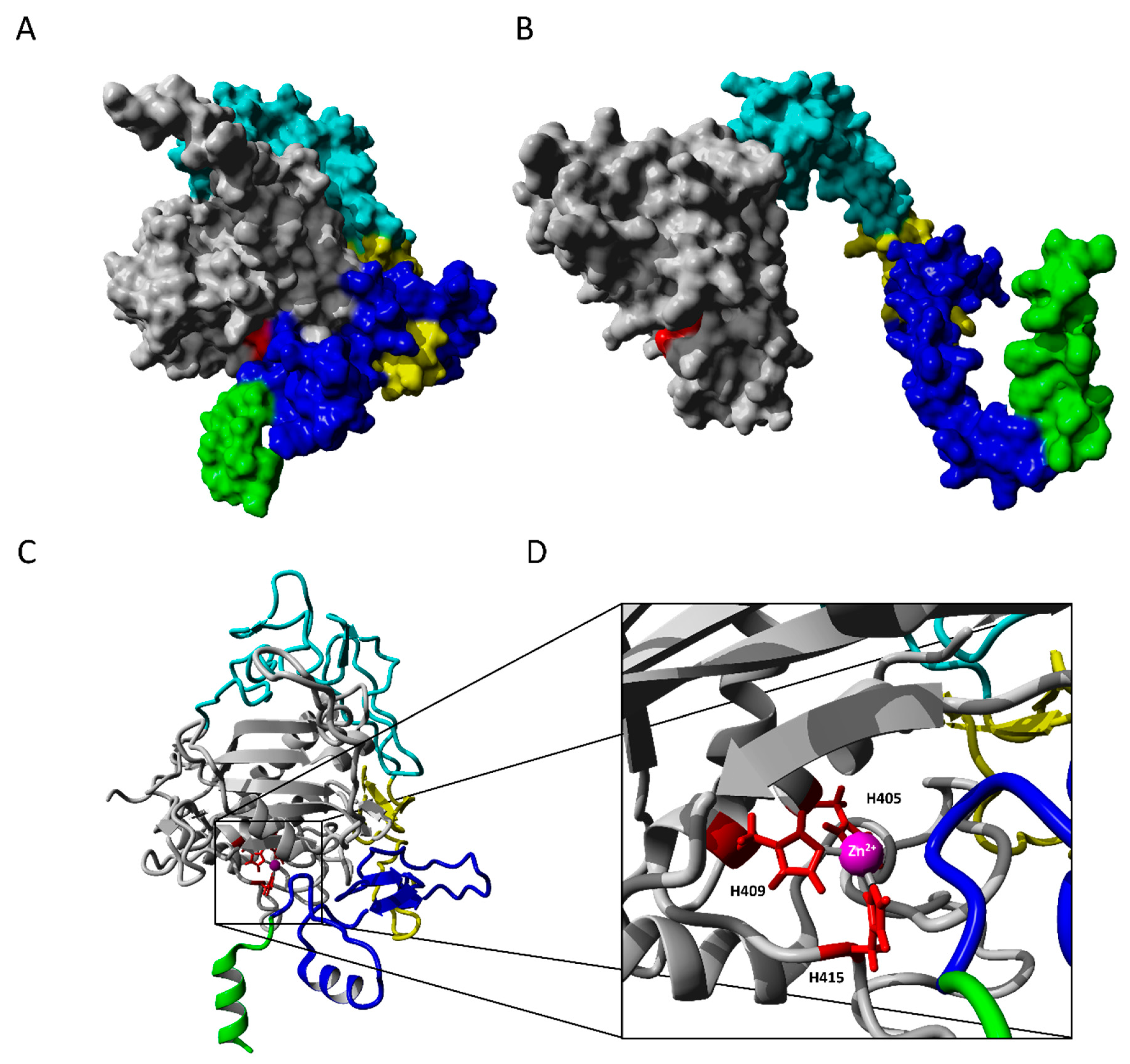
2.2.1. ADAM17 Structure Modeling
2.2.2. Molecular Docking
2.2.3. TAPI-1 and Catalytic Domain
2.2.4. Substrate II, Extracellular and Catalytic Domain of ADAM17
2.2.5. Exosite Inhibitor and the Extracellular Domain of ADAM17
3. Discussion
4. Materials and Methods
4.1. Experimental Pharmacology
4.1.1. Cell Culture, Sample Preparation and Cleavage Assay
4.1.2. Elisa Measurement
4.1.3. Western Blot
4.1.4. Expression and Purification of 1ST3
4.2. 3D Protein Structure Modeling and Molecular Docking
4.2.1. Structural Analysis
4.2.2. MD Simulation
4.2.3. Molecular Docking
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Term |
| ADAMs | A disintegrin and metalloproteinases |
| Ac-REEDANS-VHHQKLVF-KDABCYL-R-OH | Alpha Secretase Substrate II |
| AA | Amino acid |
| APP | amyloid precursor protein |
| BCA | Bicinchoninic acid |
| CID17 | Exosite inhibitor |
| GI | GI254023X |
| IPF | Idiopathic pulmonary fibrosis |
| LGA | Lamarckian Genetic Algorithm |
| MP | Metalloproteinase |
| NCBI | National Center for Biotechnology Information |
| PrAMA | Proteolytic Activity Matrix Analysis |
| TACE | Tumor necrosis factor-α converting enzyme |
| TNF | Tumor necrosis factor-α |
| YASARA | Yet Another Scientific Artificial Reality Application |
References
- Dusterhoft, S.; Michalek, M.; Kordowski, F.; Oldefest, M.; Sommer, A.; Roseler, J.; Reiss, K.; Grotzinger, J.; Lorenzen, I. Extracellular Juxtamembrane Segment of ADAM17 Interacts with Membranes and Is Essential for Its Shedding Activity. Biochemistry 2015, 54, 5791–5801. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Shen, M.; Fernandez-Patron, C.; Kassiri, Z. ADAMs family and relatives in cardiovascular physiology and pathology. J. Mol. Cell. Cardiol. 2016, 93, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D.; de Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lubke, T.; Lena Illert, A.; von Figura, K.; et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Takaishi, H.; Okada, Y.; Toyama, Y.; Blobel, C.P. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 2007, 179, 2686–2689. [Google Scholar] [CrossRef] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; Becherer, J.D. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef]
- Kwok, H.F.; Botkjaer, K.A.; Tape, C.J.; Huang, Y.; McCafferty, J.; Murphy, G. Development of a ‘mouse and human cross-reactive’ affinity-matured exosite inhibitory human antibody specific to TACE (ADAM17) for cancer immunotherapy. Protein Eng. Des. Sel. PEDS 2014, 27, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Dreymueller, D.; Ludwig, A. Considerations on inhibition approaches for proinflammatory functions of ADAM proteases. Platelets 2017, 28, 354–361. [Google Scholar] [CrossRef]
- Camodeca, C.; Nuti, E.; Tepshi, L.; Boero, S.; Tuccinardi, T.; Stura, E.A.; Poggi, A.; Zocchi, M.R.; Rossello, A. Discovery of a new selective inhibitor of A Disintegrin And Metalloprotease 10 (ADAM-10) able to reduce the shedding of NKG2D ligands in Hodgkin’s lymphoma cell models. Eur. J. Med. Chem. 2016, 111, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Seipold, L.; Saftig, P. The metalloproteinase ADAM10: A useful therapeutic target? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2071–2081. [Google Scholar] [CrossRef]
- Knapinska, A.M.; Dreymuller, D.; Ludwig, A.; Smith, L.; Golubkov, V.; Sohail, A.; Fridman, R.; Giulianotti, M.; LaVoi, T.M.; Houghten, R.A.; et al. SAR Studies of Exosite-Binding Substrate-Selective Inhibitors of A Disintegrin And Metalloprotease 17 (ADAM17) and Application as Selective in Vitro Probes. J. Med. Chem. 2015, 58, 5808–5824. [Google Scholar] [CrossRef] [Green Version]
- Madoux, F.; Dreymuller, D.; Pettiloud, J.P.; Santos, R.; Becker-Pauly, C.; Ludwig, A.; Fields, G.B.; Bannister, T.; Spicer, T.P.; Cudic, M.; et al. Discovery of an enzyme and substrate selective inhibitor of ADAM10 using an exosite-binding glycosylated substrate. Sci. Rep. 2016, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. A practical guide to modelling enzyme-catalysed reactions. Chem. Soc. Rev. 2012, 41, 3025–3038. [Google Scholar] [CrossRef]
- Fiorillo, B.; Marchiano, S.; Moraca, F.; Sepe, V.; Carino, A.; Rapacciuolo, P.; Biagioli, M.; Limongelli, V.; Zampella, A.; Catalanotti, B.; et al. Discovery of Bile Acid Derivatives as Potent ACE2 Activators by Virtual Screening and Essential Dynamics. J. Chem. Inf. Model. 2021, 62, 196–209. [Google Scholar] [CrossRef]
- Coldren, W.H.; Tikunova, S.B.; Davis, J.P.; Lindert, S. Discovery of Novel Small-Molecule Calcium Sensitizers for Cardiac Troponin C: A Combined Virtual and Experimental Screening Approach. J. Chem. Inf. Model. 2020, 60, 3648–3661. [Google Scholar] [CrossRef]
- Xue, W.; Yang, F.; Wang, P.; Zheng, G.; Chen, Y.; Yao, X.; Zhu, F. What Contributes to Serotonin-Norepinephrine Reuptake Inhibitors’ Dual-Targeting Mechanism? The Key Role of Transmembrane Domain 6 in Human Serotonin and Norepinephrine Transporters Revealed by Molecular Dynamics Simulation. ACS Chem. Neurosci. 2018, 9, 1128–1140. [Google Scholar] [CrossRef]
- Anighoro, A.; Graziani, D.; Bettinelli, I.; Cilia, A.; De Toma, C.; Longhi, M.; Mangiarotti, F.; Menegon, S.; Pirona, L.; Poggesi, E.; et al. Insights into the interaction of negative allosteric modulators with the metabotropic glutamate receptor 5: Discovery and computational modeling of a new series of ligands with nanomolar affinity. Bioorganic. Med. Chem. 2015, 23, 3040–3058. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, A.M. Flexible protein-protein docking. Curr. Opin. Struct. Biol. 2006, 16, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.J. High-resolution protein-protein docking. Curr. Opin. Struct. Biol. 2006, 16, 183–193. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Meng, S.; An, R.; Li, Z.; Schwaneberg, U.; Ji, Y.; Davari, M.D.; Wang, F.; Wang, M.; Qin, M.; Nie, K.; et al. Tunnel engineering for modulating the substrate preference in cytochrome P450BsβHI. Bioresour. Bioprocess. 2021, 8, 26. [Google Scholar] [CrossRef]
- Arkhipov, D.V.; Lomin, S.N.; Myakushina, Y.A.; Savelieva, E.M.; Osolodkin, D.I.; Romanov, G.A. Modeling of Protein-Protein Interactions in Cytokinin Signal Transduction. Int. J. Mol. Sci. 2019, 20, 2096. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C. Beware of docking! Trends Pharm. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Thiele, M.J.; Davari, M.D.; König, M.; Hofmann, I.; Junker, N.O.; Mirzaei Garakani, T.; Vojcic, L.; Fitter, J.; Schwaneberg, U. Enzyme–Polyelectrolyte Complexes Boost the Catalytic Performance of Enzymes. ACS Catal. 2018, 8, 10876–10887. [Google Scholar] [CrossRef]
- Goddette, D.W.; Paech, C.; Yang, S.S.; Mielenz, J.R.; Bystroff, C.; Wilke, M.E.; Fletterick, R.J. The crystal structure of the Bacillus lentus alkaline protease, subtilisin BL, at 1.4 Å resolution. J. Mol. Biol. 1992, 228, 580–595. [Google Scholar] [CrossRef]
- Minond, D.; Cudic, M.; Bionda, N.; Giulianotti, M.; Maida, L.; Houghten, R.A.; Fields, G.B. Discovery of novel inhibitors of a disintegrin and metalloprotease 17 (ADAM17) using glycosylated and non-glycosylated substrates. J. Biol. Chem. 2012, 287, 36473–36487. [Google Scholar] [CrossRef] [Green Version]
- Groth, E. Regulation und Transport der Disintegrin und Metalloproteinasen ADAM10 und ADAM17. RWTH Aachen, Aachen, 2016. Available online: https://bioresourcesbioprocessing.springeropen.com/articles/10.1186/s40643-021-00379 (accessed on 11 January 2022).
- Pruessmeyer, J.; Hess, F.M.; Alert, H.; Groth, E.; Pasqualon, T.; Schwarz, N.; Nyamoya, S.; Kollert, J.; van der Vorst, E.; Donners, M. Leukocytes require ADAM10 but not ADAM17 for their migration and inflammatory recruitment into the alveolar space. Blood J. Am. Soc. Hematol. 2014, 123, 4077–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selner, N.G.; Luechapanichkul, R.; Chen, X.; Neel, B.G.; Zhang, Z.-Y.; Knapp, S.; Bell, C.E.; Pei, D. Diverse Levels of Sequence Selectivity and Catalytic Efficiency of Protein-Tyrosine Phosphatases. Biochemistry 2014, 53, 397–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, C.; Miller, M.A.; Bartsch, J.W.; Schlomann, U.; Lauffenburger, D.A. Simultaneous Detection of Metalloprotease Activities in Complex Biological Samples Using the PrAMA (Proteolytic Activity Matrix Assay) Method. Methods Mol. Biol. 2017, 1574, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Maskos, K.; Fernandez-Catalan, C.; Huber, R.; Bourenkov, G.P.; Bartunik, H.; Ellestad, G.A.; Reddy, P.; Wolfson, M.F.; Rauch, C.T.; Castner, B.J.; et al. Crystal structure of the catalytic domain of human tumor necrosis factor-alpha-converting enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 3408–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, R.N.; Orth, P.; Strickland, C.L.; Le, H.V.; Madison, V.; Beyer, B.M. Stabilization of the autoproteolysis of TNF-alpha converting enzyme (TACE) results in a novel crystal form suitable for structure-based drug design studies. Protein Eng. Des. Sel. PEDS 2006, 19, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.A.; Jiao, G.S.; Kim, S.; Thai, A.; Cregar-Hernandez, L.; Margosiak, S.A.; Johnson, A.T.; Han, G.W.; O’Malley, S.; Stevens, R.C. Structural characterization of three novel hydroxamate-based zinc chelating inhibitors of the Clostridium botulinum serotype A neurotoxin light chain metalloprotease reveals a compact binding site resulting from 60/70 loop flexibility. Biochemistry 2011, 50, 4019–4028. [Google Scholar] [CrossRef] [Green Version]
- Laronha, H.; Carpinteiro, I.; Portugal, J.; Azul, A.; Polido, M.; Petrova, K.T.; Salema-Oom, M.; Caldeira, J. Challenges in Matrix Metalloproteinases Inhibition. Biomolecules 2020, 10, 717. [Google Scholar] [CrossRef]
- Song, J.; Tan, H.; Perry, A.J.; Akutsu, T.; Webb, G.I.; Whisstock, J.C.; Pike, R.N. PROSPER: An integrated feature-based tool for predicting protease substrate cleavage sites. PLoS ONE 2012, 7, e50300. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Liu, J.; Obando, D.; Liao, V.; Lifa, T.; Codd, R. The many faces of the adamantyl group in drug design. Eur. J. Med. Chem. 2011, 46, 1949–1963. [Google Scholar] [CrossRef] [PubMed]
- Mullberg, J.; Durie, F.H.; Otten-Evans, C.; Alderson, M.R.; Rose-John, S.; Cosman, D.; Black, R.A.; Mohler, K.M. A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J. Immunol. 1995, 155, 5198–5205. [Google Scholar] [PubMed]
- Fernandez-Real, J.M.; Lainez, B.; Vendrell, J.; Rigla, M.; Castro, A.; Penarroja, G.; Broch, M.; Perez, A.; Richart, C.; Engel, P.; et al. Shedding of TNF-alpha receptors, blood pressure, and insulin sensitivity in type 2 diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E952–E959. [Google Scholar] [CrossRef] [PubMed]
- Xanthoulea, S.; Pasparakis, M.; Kousteni, S.; Brakebusch, C.; Wallach, D.; Bauer, J.; Lassmann, H.; Kollias, G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 2004, 200, 367–376. [Google Scholar] [CrossRef]
- Bartsch, J.W.; Wildeboer, D.; Koller, G.; Naus, S.; Rittger, A.; Moss, M.L.; Minai, Y.; Jockusch, H. Tumor necrosis factor-α (TNF-α) regulates shedding of TNF-α receptor 1 by the metalloprotease-disintegrin ADAM8: Evidence for a protease-regulated feedback loop in neuroprotection. J. Neurosci. 2010, 30, 12210–12218. [Google Scholar] [CrossRef]
- Dennis, M.S.; Eigenbrot, C.; Skelton, N.J.; Ultsch, M.H.; Santell, L.; Dwyer, M.A.; O’Connell, M.P.; Lazarus, R.A. Peptide exosite inhibitors of factor VIIa as anticoagulants. Nature 2000, 404, 465–470. [Google Scholar] [CrossRef]
- Roth, J.; Minond, D.; Darout, E.; Liu, Q.; Lauer, J.; Hodder, P.; Fields, G.B.; Roush, W.R. Identification of novel, exosite-binding matrix metalloproteinase-13 inhibitor scaffolds. Bioorg. Med. Chem. Lett. 2011, 21, 7180–7184. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. YASARA View-molecular graphics for all devices-from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Tape, C.J.; Willems, S.H.; Dombernowsky, S.L.; Stanley, P.L.; Fogarasi, M.; Ouwehand, W.; McCafferty, J.; Murphy, G. Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. USA 2011, 108, 5578–5583. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S.; Chalaris, A. ADAM17: A potential therapeutic target for rheumatoid arthritis? Int. J. Clin. Rheumatol. 2012, 7, 357. [Google Scholar] [CrossRef]
- Umemura, M.; Isozaki, T.; Ishii, S.; Seki, S.; Oguro, N.; Miura, Y.; Miwa, Y.; Nakamura, M.; Inagaki, K.; Kasama, T. Reduction of Serum ADAM17 Level Accompanied with Decreased Cytokines after Abatacept Therapy in Patients with Rheumatoid Arthritis. Int. J. Biomed. Sci. 2014, 10, 229–235. [Google Scholar] [PubMed]
- Arribas, J.; Esselens, C. ADAM17 as a therapeutic target in multiple diseases. Curr. Pharm. Des. 2009, 15, 2319–2335. [Google Scholar] [CrossRef] [PubMed]
- Shou, Z.X.; Jin, X.; Zhao, Z.S. Upregulated expression of ADAM17 is a prognostic marker for patients with gastric cancer. Ann. Surg. 2012, 256, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.I.; Rose-John, S.; Jenkins, B.J. ADAM17: An Emerging Therapeutic Target for Lung Cancer. Cancers 2019, 11, 1218. [Google Scholar] [CrossRef] [Green Version]
- Palau, V.; Riera, M.; Soler, M.J. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol. Dial. Transplant. 2020, 35, 1071–1072. [Google Scholar] [CrossRef]
- Stepanova, G. Biologia Futura: Is ADAM 17 the reason for COVID-19 susceptibility in hyperglycemic and diabetic patients? Biol. Futur. 2021, 72, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Palacios, Y.; Ruiz, A.; Ramon-Luing, L.A.; Ocana-Guzman, R.; Barreto-Rodriguez, O.; Sanchez-Moncivais, A.; Tecuatzi-Cadena, B.; Regalado-Garcia, A.G.; Pineda-Gudino, R.D.; Garcia-Martinez, A.; et al. Severe COVID-19 Patients Show an Increase in Soluble TNFR1 and ADAM17, with a Relationship to Mortality. Int. J. Mol. Sci. 2021, 22, 8423. [Google Scholar] [CrossRef]
- Schreiber, B.; Patel, A.; Verma, A. Shedding Light on COVID-19: ADAM17 the Missing Link? Am. J. Ther. 2020, 28, e358–e360. [Google Scholar] [CrossRef]
- Zipeto, D.; Palmeira, J.D.F.; Arganaraz, G.A.; Arganaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 11, 576745. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Rosner, K.E.; Guo, Z.; Orth, P.; Shipps, G.W., Jr.; Belanger, D.B.; Chan, T.Y.; Curran, P.J.; Dai, C.; Deng, Y.; Girijavallabhan, V.M.; et al. The discovery of novel tartrate-based TNF-alpha converting enzyme (TACE) inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Guo, Z.; Orth, P.; Madison, V.; Chen, L.; Dai, C.; Feltz, R.J.; Girijavallabhan, V.M.; Kim, S.H.; Kozlowski, J.A.; et al. Discovery and SAR of hydantoin TACE inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1877–1880. [Google Scholar] [CrossRef]
- Yu, W.; Tong, L.; Kim, S.H.; Wong, M.K.; Chen, L.; Yang, D.Y.; Shankar, B.B.; Lavey, B.J.; Zhou, G.; Kosinski, A.; et al. Biaryl substituted hydantoin compounds as TACE inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 5286–5289. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Popovici-Muller, J.; Belanger, D.B.; Caldwell, J.; Dai, C.; David, M.; Girijavallabhan, V.M.; Lavey, B.J.; Lee, J.F.; Liu, Z.; et al. Structure and activity relationships of tartrate-based TACE inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 4812–4815. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Li, D.; Popovici-Muller, J.; Zhao, L.; Girijavallabhan, V.M.; Rosner, K.E.; Lavey, B.J.; Rizvi, R.; Shankar, B.B.; Wong, M.K.; et al. 2-(2-Aminothiazol-4-yl)pyrrolidine-based tartrate diamides as potent, selective and orally bioavailable TACE inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3172–3176. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Gopalsamy, A.; Aplasca, A.; Ellingboe, J.W.; Xu, W.; Zhang, Y.; Levin, J.I. Synthesis and activity of tryptophan sulfonamide derivatives as novel non-hydroxamate TNF-alpha converting enzyme (TACE) inhibitors. Bioorganic Med. Chem. 2009, 17, 3857–3865. [Google Scholar] [CrossRef]
- Mazzola, R.D., Jr.; Zhu, Z.; Sinning, L.; McKittrick, B.; Lavey, B.; Spitler, J.; Kozlowski, J.; Neng-Yang, S.; Zhou, G.; Guo, Z.; et al. Discovery of novel hydroxamates as highly potent tumor necrosis factor-alpha converting enzyme inhibitors. Part II: Optimization of the S3’ pocket. Bioorg. Med. Chem. Lett. 2008, 18, 5809–5814. [Google Scholar] [CrossRef]
- Guo, Z.; Orth, P.; Wong, S.C.; Lavey, B.J.; Shih, N.Y.; Niu, X.; Lundell, D.J.; Madison, V.; Kozlowski, J.A. Discovery of novel spirocyclopropyl hydroxamate and carboxylate compounds as TACE inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 54–57. [Google Scholar] [CrossRef]
- Bandarage, U.K.; Wang, T.; Come, J.H.; Perola, E.; Wei, Y.; Rao, B.G. Novel thiol-based TACE inhibitors. Part 2: Rational design, synthesis, and SAR of thiol-containing aryl sulfones. Bioorg. Med. Chem. Lett. 2008, 18, 44–48. [Google Scholar] [CrossRef]
- Levin, J.I.; Chen, J.M.; Laakso, L.M.; Du, M.; Du, X.; Venkatesan, A.M.; Sandanayaka, V.; Zask, A.; Xu, J.; Xu, W.; et al. Acetylenic TACE inhibitors. Part 2: SAR of six-membered cyclic sulfonamide hydroxamates. Bioorg. Med. Chem. Lett. 2005, 15, 4345–4349. [Google Scholar] [CrossRef]
- Levin, J.I.; Chen, J.M.; Laakso, L.M.; Du, M.; Schmid, J.; Xu, W.; Cummons, T.; Xu, J.; Jin, G.; Barone, D.; et al. Acetylenic TACE inhibitors. Part 3: Thiomorpholine sulfonamide hydroxamates. Bioorg. Med. Chem. Lett. 2006, 16, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Condon, J.S.; Joseph-McCarthy, D.; Levin, J.I.; Lombart, H.G.; Lovering, F.E.; Sun, L.; Wang, W.; Xu, W.; Zhang, Y. Identification of potent and selective TACE inhibitors via the S1 pocket. Bioorg. Med. Chem. Lett. 2007, 17, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Umland, S.; Ingram, R.; Beyer, B.M.; Liu, Y.H.; Sun, J.; Lundell, D.; Orth, P. IK682, a tight binding inhibitor of TACE. Arch. Biochem. Biophys. 2006, 451, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. 9), 114–122. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham III, T.; Darden, T.; Duke, R.; Gohlke, H. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, G.; Huang, X.; Black, R.; Wolfson, M.; Rauch, C.; McGregor, H.; Ellestad, G.; Cowling, R. A continuous fluorimetric assay for tumor necrosis factor-alpha converting enzyme. Anal. Biochem. 2002, 302, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4(Zn): An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bienstein, M.; Minond, D.; Schwaneberg, U.; Davari, M.D.; Yildiz, D. In Silico and Experimental ADAM17 Kinetic Modeling as Basis for Future Screening System for Modulators. Int. J. Mol. Sci. 2022, 23, 1368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031368
Bienstein M, Minond D, Schwaneberg U, Davari MD, Yildiz D. In Silico and Experimental ADAM17 Kinetic Modeling as Basis for Future Screening System for Modulators. International Journal of Molecular Sciences. 2022; 23(3):1368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031368
Chicago/Turabian StyleBienstein, Marian, Dmitriy Minond, Ulrich Schwaneberg, Mehdi D. Davari, and Daniela Yildiz. 2022. "In Silico and Experimental ADAM17 Kinetic Modeling as Basis for Future Screening System for Modulators" International Journal of Molecular Sciences 23, no. 3: 1368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031368