
To Be or Not to Be: The Divergent Action and Metabolism of Sphingosine-1 Phosphate in Pancreatic Beta-Cells in Response to Cytokines and Fatty Acids


Abstract
:1. Introduction
2. Overview of Mechanisms of Beta-Cell Destruction in T1DM
3. Overview of Mechanisms of Beta-Cell Destruction in T2DM
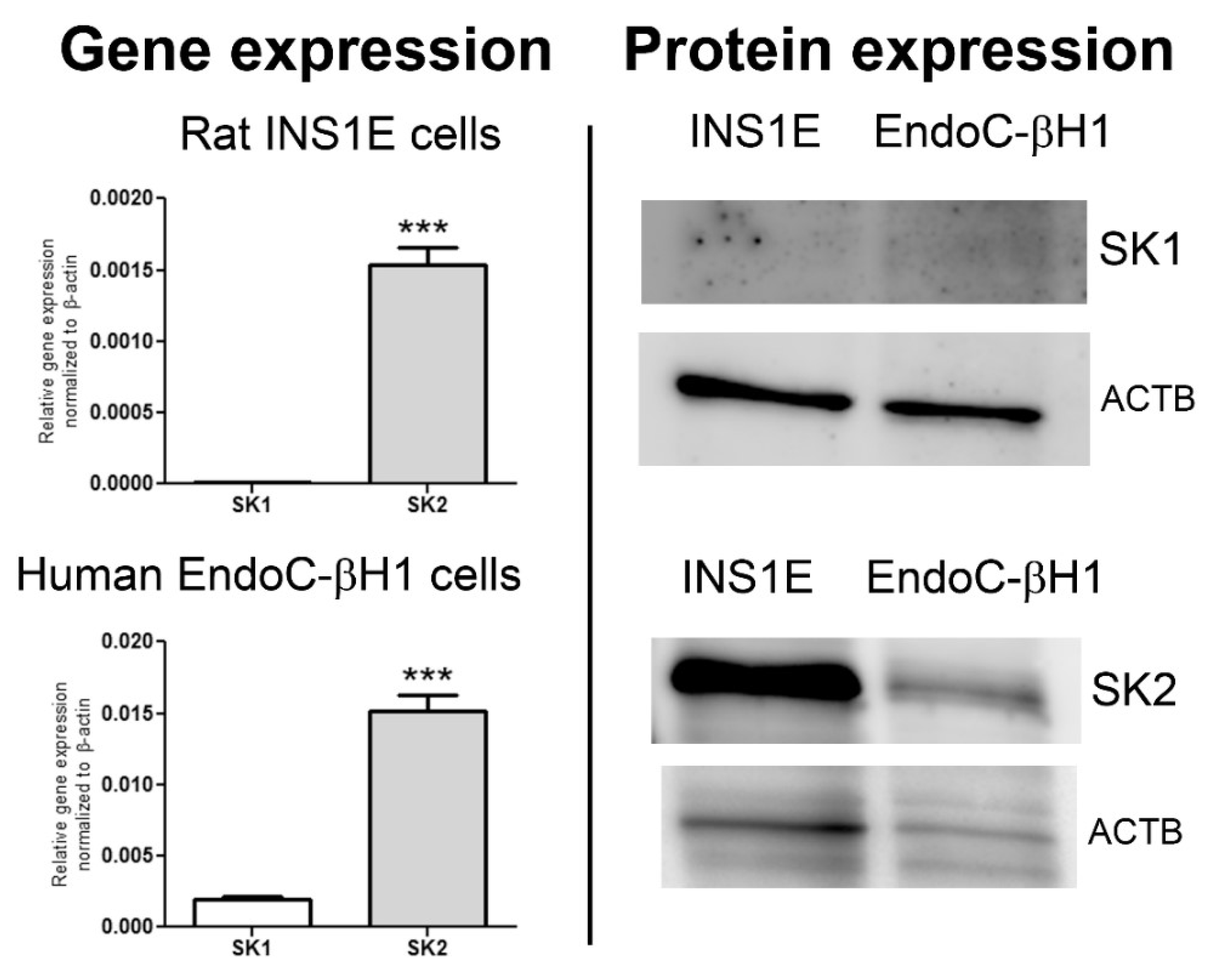
4. Sphingosine-1 Phosphate Metabolism, Receptors and Transporters in Pancreatic Beta-Cells
5. Effects of Proinflammatory Cytokines on S1P Metabolism in Pancreatic Beta-Cells
6. Effects of Fatty Acids and Hyperglycemia on S1P Metabolism in Pancreatic Beta-Cells
7. Effects of S1P on Cytokine Toxicity in Pancreatic Beta-Cells
7.1. Extracellular S1P
7.2. Intracellular S1P
8. Effects of S1P on FFA Toxicity in Pancreatic Beta-Cells
8.1. Extracellular S1P
8.2. Intracellular S1P
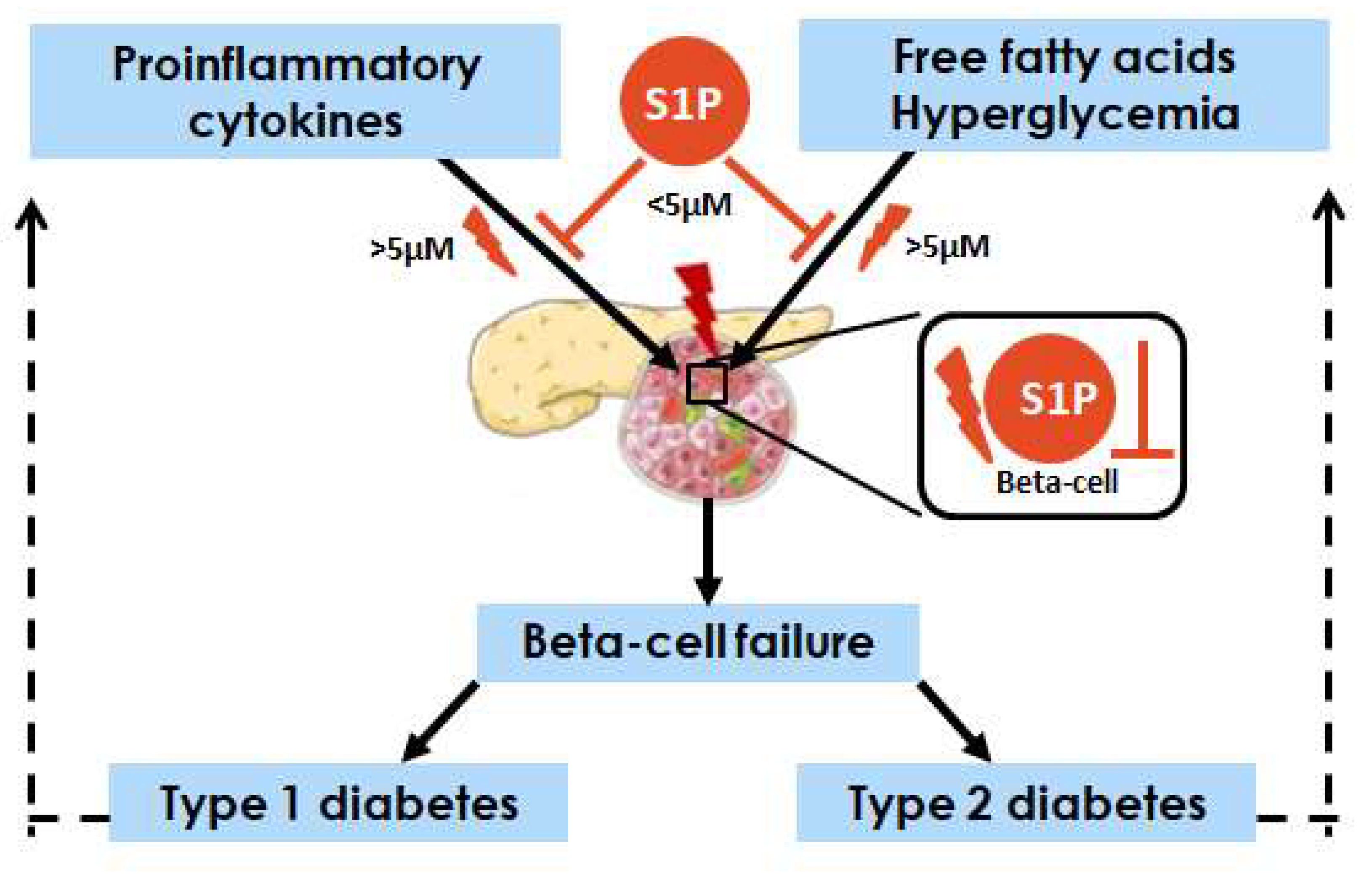
9. Similarities and Differences in S1P Action in Beta-Cells under T1DM and T2DM
10. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franks, P.W.; Merino, J. Gene-lifestyle interplay in type 2 diabetes. Curr. Opin. Genet. Dev. 2018, 50, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107. [Google Scholar] [CrossRef] [Green Version]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.J.; Krogvold, L.; Hasselby, J.P.; Kaur, S.; Claessens, L.A.; Russell, M.A.; Mathews, C.E.; Hanssen, K.F.; Morgan, N.G.; Koeleman, B.P.C.; et al. Abnormal islet sphingolipid metabolism in type 1 diabetes. Diabetologia 2018, 61, 1650–1661. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Hla, T.; Dannenberg, A.J. Sphingolipid signaling in metabolic disorders. Cell Metab. 2012, 16, 420–434. [Google Scholar] [CrossRef] [Green Version]
- Jessup, C.F.; Bonder, C.S.; Pitson, S.M.; Coates, P.T. The sphingolipid rheostat: A potential target for improving pancreatic islet survival and function. Endocr. Metab. Immune Disord. Drug Targets 2011, 11, 262–272. [Google Scholar] [CrossRef]
- Veret, J.; Bellini, L.; Giussani, P.; Ng, C.; Magnan, C.; Le Stunff, H. Roles of sphingolipid metabolism in pancreatic beta cell dysfunction induced by lipotoxicity. J. Clin. Med. 2014, 3, 646–662. [Google Scholar] [CrossRef] [Green Version]
- Oresic, M.; Simell, S.; Sysi-Aho, M.; Nanto-Salonen, K.; Seppanen-Laakso, T.; Parikka, V.; Katajamaa, M.; Hekkala, A.; Mattila, I.; Keskinen, P.; et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J. Exp. Med. 2008, 205, 2975–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, N.; Pan, J.; Pop-Busui, R.; Othman, A.; Alecu, I.; Hornemann, T.; Eichler, F.S. Altered sphingoid base profiles in type 1 compared to type 2 diabetes. Lipids Health Dis. 2014, 13, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, T.E.; Bewley, M.C.; Unrath, K.A.; Pedersen, M.M.; Anderson, R.E.; Jung, D.Y.; Jefferson, L.S.; Kim, J.K.; Bronson, S.K.; Flanagan, J.M.; et al. Circulating sphingolipid biomarkers in models of type 1 diabetes. J. Lipid Res. 2011, 52, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Dickens, A.M.; Lopez-Bascon, M.A.; Lindeman, T.; Kemppainen, E.; Lamichhane, S.; Ronkko, T.; Ilonen, J.; Toppari, J.; Veijola, R.; et al. Metabolic alterations in immune cells associate with progression to type 1 diabetes. Diabetologia 2020, 63, 1017–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate: The Swiss army knife of sphingolipid signaling. J. Lipid Res. 2009, 50, S272–S276. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, D.L.; Fu, P.; Ramchandran, R.; Ha, A.W.; Putherickal, V.; Sudhadevi, T.; Harijith, A.; Schumacher, F.; Kleuser, B.; Natarajan, V. S1P and plasmalogen derived fatty aldehydes in cellular signaling and functions. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2020, 1865, 158681. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Ebenezer, D.L.; Ha, A.W.; Suryadevara, V.; Harijith, A.; Natarajan, V. Nuclear lipid mediators: Role of nuclear sphingolipids and sphingosine-1-phosphate signaling in epigenetic regulation of inflammation and gene expression. J. Cell Biochem. 2018, 119, 6337–6353. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, N.M.; Longacre, M.J.; Stoker, S.W.; Kendrick, M.A.; Druckenbrod, N.R.; Laychock, S.G.; Mastrandrea, L.D.; MacDonald, M.J. Sphingosine kinase 1 knockdown reduces insulin synthesis and secretion in a rat insulinoma cell line. Arch. Biochem. Biophys. 2012, 518, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Veret, J.; Coant, N.; Gorshkova, I.A.; Giussani, P.; Fradet, M.; Riccitelli, E.; Skobeleva, A.; Goya, J.; Kassis, N.; Natarajan, V.; et al. Role of palmitate-induced sphingoid base-1-phosphate biosynthesis in INS-1 beta-cell survival. Biochim. Biophys. Acta 2013, 1831, 251–262. [Google Scholar] [CrossRef]
- Japtok, L.; Schmitz, E.I.; Fayyaz, S.; Kramer, S.; Hsu, L.J.; Kleuser, B. Sphingosine 1-phosphate counteracts insulin signaling in pancreatic beta-cells via the sphingosine 1-phosphate receptor subtype 2. FASEB J. 2015, 29, 3357–3369. [Google Scholar] [CrossRef] [Green Version]
- Cantrell Stanford, J.; Morris, A.J.; Sunkara, M.; Popa, G.J.; Larson, K.L.; Ozcan, S. Sphingosine 1-phosphate (S1P) regulates glucose-stimulated insulin secretion in pancreatic beta cells. J. Biol. Chem. 2012, 287, 13457–13464. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Chen, J.; Lay, A.; Don, A.; Vadas, M.; Xia, P. Loss of sphingosine kinase 1 predisposes to the onset of diabetes via promoting pancreatic beta-cell death in diet-induced obese mice. FASEB J. 2013, 27, 4294–4304. [Google Scholar] [CrossRef]
- Taguchi, Y.; Allende, M.L.; Mizukami, H.; Cook, E.K.; Gavrilova, O.; Tuymetova, G.; Clarke, B.A.; Chen, W.; Olivera, A.; Proia, R.L. Sphingosine-1-phosphate phosphatase 2 regulates pancreatic islet beta-cell endoplasmic reticulum stress and proliferation. J. Biol. Chem. 2016, 291, 12029–12038. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.; Tyka, K.; Saba, J.D.; Lenzen, S.; Gurgul-Convey, E. Overexpression of sphingosine-1-phosphate lyase protects insulin-secreting cells against cytokine toxicity. J. Biol. Chem. 2017, 292, 20292–20304. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Plötz, T.; Gräler, M.H.; Gurgul-Convey, E. Sphingosine-1 phosphate lyase regulates sensitivity of pancreatic beta-cells to lipotoxicity. Int. J. Mol. Sci. 2021, 22, 10893. [Google Scholar] [CrossRef] [PubMed]
- Roep, B.O.; Peakman, M. Diabetogenic T lymphocytes in human type 1 diabetes. Curr. Opin. Immunol. 2011, 23, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Lernmark, A. 2016. Genetic risk factors for type 1 diabetes. Lancet 2021, 387, 2331–2339. [Google Scholar] [CrossRef]
- Coppieters, K.T.; von Herrath, M.G. The type 1 diabetes signature: Hardwired to trigger inflammation? Diabetes 2014, 63, 3581–3583. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Colli, M.L.; Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009, 5, 219–226. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Suarez-Pinzon, W.L. Role of cytokines in the pathogenesis of autoimmune diabetes mellitus. Rev. Endocr. Metab. Disord. 2003, 4, 291–299. [Google Scholar] [CrossRef]
- Andersen, N.A.; Larsen, C.M.; Mandrup-Poulsen, T. TNFalpha and IFNgamma potentiate IL-1beta induced mitogen activated protein kinase activity in rat pancreatic islets of Langerhans. Diabetologia 2000, 43, 1389–1396. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Mandrup-Poulsen, T. A choice of death-the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 2001, 44, 2115–2133. [Google Scholar] [CrossRef]
- Hansen, J.B.; Moen, I.W.; Mandrup-Poulsen, T. Iron: The hard player in diabetes pathophysiology. Acta Physiol. 2014, 210, 717–732. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Grieco, F.A. On the immense variety and complexity of circumstances conditioning pancreatic beta-cell apoptosis in type 1 diabetes. Diabetes 2012, 61, 1661–1663. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef] [PubMed]
- Villate, O.; Turatsinze, J.V.; Mascali, L.G.; Grieco, F.A.; Nogueira, T.C.; Cunha, D.A.; Nardelli, T.R.; Sammeth, M.; Salunkhe, V.A.; Esguerra, J.L.; et al. Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucleic Acids Res. 2014, 42, 11818–11830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demine, S.; Schiavo, A.A.; Marin-Canas, S.; Marchetti, P.; Cnop, M.; Eizirik, D.L. Pro-inflammatory cytokines induce cell death, inflammatory responses, and endoplasmic reticulum stress in human iPSC-derived beta cells. Stem Cell Res. Ther. 2020, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, K.L.; Gurgul-Convey, E.; Elsner, M.; Lenzen, S. Interaction between pro-inflammatory and anti-inflammatory cytokines in insulin-producing cells. J. Endocrinol. 2008, 197, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurgul-Convey, E.; Lenzen, S. Protection against cytokine toxicity through endoplasmic reticulum and mitochondrial stress prevention by prostacyclin synthase overexpression in insulin-producing cells. J. Biol. Chem. 2010, 285, 11121–11128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurgul-Convey, E.; Mehmeti, I.; Lortz, S.; Lenzen, S. Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J. Mol. Med. 2011, 89, 785–798. [Google Scholar] [CrossRef]
- Kacheva, S.; Lenzen, S.; Gurgul-Convey, E. Differential effects of proinflammatory cytokines on cell death and ER stress in insulin-secreting INS1E cells and the involvement of nitric oxide. Cytokine 2011, 55, 195–201. [Google Scholar] [CrossRef]
- Mehmeti, I.; Gurgul-Convey, E.; Lenzen, S.; Lortz, S. Induction of the intrinsic apoptosis pathway in insulin-secreting cells is dependent on oxidative damage of mitochondria but independent of caspase-12 activation. Biochim. Biophys. Acta 2011, 1813, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Hanzelka, K.; Skalniak, L.; Jura, J.; Lenzen, S.; Gurgul-Convey, E. Effects of the novel mitochondrial protein mimitin in insulin-secreting cells. Biochem. J. 2012, 445, 349–359. [Google Scholar] [CrossRef]
- Gurgul-Convey, E.; Mehmeti, I.; Plötz, T.; Jörns, A.; Lenzen, S. Sensitivity profile of the human EndoC-betaH1 beta cell line to proinflammatory cytokines. Diabetologia 2016, 59, 2125–2133. [Google Scholar] [CrossRef] [Green Version]
- Tyka, K.; Jörns, A.; Turatsinze, J.V.; Eizirik, D.L.; Lenzen, S.; Gurgul-Convey, E. MCPIP1 regulates the sensitivity of pancreatic beta-cells to cytokine toxicity. Cell Death Dis. 2019, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, E.; Lortz, S.; Tiedge, M.; Jörns, A.; Lenzen, S. Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 2004, 53, 2271–2280. [Google Scholar] [CrossRef] [Green Version]
- Lenzen, S. Oxidative stress: The vulnerable beta-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, S.M.; Dahllof, M.S.; Osmai, Y.; Osmai, M.; Jakobsen, K.K.; Aivazidis, A.; Tyrberg, B.; Perruzza, L.; Prause, M.C.B.; Christensen, D.P.; et al. Regulation of the beta-cell inflammasome and contribution to stress-induced cellular dysfunction and apoptosis. Mol. Cell. Endocrinol. 2018, 478, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, S.M.; Krogh, N.; Tyrberg, B.; Mandrup-Poulsen, T. The No-Go and Nonsense-Mediated RNA Decay Pathways Are Regulated by Inflammatory Cytokines in Insulin-Producing Cells and Human Islets and Determine beta-Cell Insulin Biosynthesis and Survival. Diabetes 2018, 67, 2019–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyka, K.; Jörns, A.; Dunst, A.; Tang, Y.; Bryde, T.H.; Mehmeti, I.; Walentinsson, A.; Marselli, L.; Cnop, M.; Tyrberg, B.; et al. MCPIP1 is a novel link between diabetogenic conditions and impaired insulin secretory capacity. Biochim. Biophys. Acta. Mol. Basis Dis. 2021, 1867, 166199. [Google Scholar] [CrossRef] [PubMed]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef]
- Lortz, S.; Gurgul-Convey, E.; Naujok, O.; Lenzen, S. Overexpression of the antioxidant enzyme catalase does not interfere with the glucose responsiveness of insulin-secreting INS-1E cells and rat islets. Diabetologia 2013, 56, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, A.K.; Heimberg, H.; Heremans, Y.; Leeman, R.; Kutlu, B.; Kruhoffer, M.; Orntoft, T.; Eizirik, D.L. A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J. Biol. Chem. 2001, 276, 48879–48886. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, A.K.; Kruhoffer, M.; Leeman, R.; Orntoft, T.; Eizirik, D.L. Identification of novel cytokine-induced genes in pancreatic beta-cells by high-density oligonucleotide arrays. Diabetes 2001, 50, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, A.K.; Proost, P.; Gysemans, C.; Chen, M.C.; Mathieu, C.; Eizirik, D.L. IL-1beta and IFN-gamma induce the expression of diverse chemokines and IL-15 in human and rat pancreatic islet cells, and in islets from pre-diabetic NOD mice. Diabetologia 2003, 46, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Proost, P.; Gysemans, C.; Mathieu, C.; Eizirik, D.L. Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1 beta-exposed human and rat islet cells. Diabetologia 2001, 44, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Miani, M.; Cardozo, A.K. Signalling danger: Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013, 56, 234–241. [Google Scholar] [CrossRef]
- Jacobsen, M.L.; Ronn, S.G.; Bruun, C.; Larsen, C.M.; Eizirik, D.L.; Mandrup-Poulsen, T.; Billestrup, N. IL-1beta-induced chemokine and Fas expression are inhibited by suppressor of cytokine signalling-3 in insulin-producing cells. Diabetologia 2009, 52, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Kutlu, B.; Cardozo, A.K.; Darville, M.I.; Kruhoffer, M.; Magnusson, N.; Orntoft, T.; Eizirik, D.L. Discovery of gene networks regulating cytokine-induced dysfunction and apoptosis in insulin-producing INS-1 cells. Diabetes 2003, 52, 2701–2719. [Google Scholar] [CrossRef] [Green Version]
- Kutlu, B.; Darville, M.I.; Cardozo, A.K.; Eizirik, D.L. Molecular regulation of monocyte chemoattractant protein-1 expression in pancreatic beta-cells. Diabetes 2003, 52, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Miani, M.; Colli, M.L.; Ladriere, L.; Cnop, M.; Eizirik, D.L. Mild endoplasmic reticulum stress augments the proinflammatory effect of IL-1beta in pancreatic rat beta-cells via the IRE1alpha/XBP1s pathway. Endocrinology 2012, 153, 3017–3028. [Google Scholar] [CrossRef]
- Moore, F.; Naamane, N.; Colli, M.L.; Bouckenooghe, T.; Ortis, F.; Gurzov, E.N.; Igoillo-Esteve, M.; Mathieu, C.; Bontempi, G.; Thykjaer, T.; et al. STAT1 is a master regulator of pancreatic {beta}-cell apoptosis and islet inflammation. J. Biol. Chem. 2011, 286, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Ortis, F.; Naamane, N.; Flamez, D.; Ladriere, L.; Moore, F.; Cunha, D.A.; Colli, M.L.; Thykjaer, T.; Thorsen, K.; Orntoft, T.F.; et al. Cytokines interleukin-1beta and tumor necrosis factor-alpha regulate different transcriptional and alternative splicing networks in primary beta-cells. Diabetes 2010, 59, 358–374. [Google Scholar] [CrossRef] [Green Version]
- Sandler, S.; Andersson, A.; Hellerström, C. Inhibitory effects of interleukin 1 on insulin secretion, insulin biosynthesis, and oxidative metabolism of isolated rat pancreatic islets. Endocrinology 1987, 121, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Southern, C.; Schulster, D.; Green, I.C. Inhibition of insulin secretion by interleukin-1 beta and tumour necrosis factor-alpha via an L-arginine-dependent nitric oxide generating mechanism. FEBS Lett. 1990, 276, 42–44. [Google Scholar] [CrossRef] [Green Version]
- Dooley, J.; Tian, L.; Schonefeldt, S.; Delghingaro-Augusto, V.; Garcia-Perez, J.E.; Pasciuto, E.; Di Marino, D.; Carr, E.J.; Oskolkov, N.; Lyssenko, V.; et al. Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat. Genet. 2016, 48, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, S.; Ahonen, L.; Dyrlund, T.S.; Dickens, A.M.; Siljander, H.; Hyoty, H.; Ilonen, J.; Toppari, J.; Veijola, R.; Hyotylainen, T.; et al. Cord-Blood Lipidome in Progression to Islet Autoimmunity and Type 1 Diabetes. Biomolecules 2019, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamichhane, S.; Kemppainen, E.; Trost, K.; Siljander, H.; Hyoty, H.; Ilonen, J.; Toppari, J.; Veijola, R.; Hyotylainen, T.; Knip, M.; et al. Circulating metabolites in progression to islet autoimmunity and type 1 diabetes. Diabetologia 2019, 62, 2287–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleich, D.; Polak, M.; Chen, S.; Swiderek, K.M.; Levy-Marchal, C. Sera from children with type 1 diabetes mellitus react against a new group of antigens composed of lysophospholipids. Horm. Res. 1999, 52, 86–94. [Google Scholar] [CrossRef]
- Jörns, A.; Akin, M.; Arndt, T.; Terbish, T.; Zu Vilsendorf, A.M.; Wedekind, D.; Hedrich, H.J.; Lenzen, S. Anti-TCR therapy combined with fingolimod for reversal of diabetic hyperglycemia by beta cell regeneration in the LEW.1AR1-iddm rat model of type 1 diabetes. J. Mol. Med. 2014, 92, 743–755. [Google Scholar]
- Jörns, A.; Rath, K.J.; Terbish, T.; Arndt, T.; Meyer Zu Vilsendorf, A.; Wedekind, D.; Hedrich, H.J.; Lenzen, S. Diabetes prevention by immunomodulatory FTY720 treatment in the LEW.1AR1-iddm rat despite immune cell activation. Endocrinology 2010, 151, 3555–3565. [Google Scholar] [CrossRef]
- Penaranda, C.; Tang, Q.; Ruddle, N.H.; Bluestone, J.A. Prevention of diabetes by FTY720-mediated stabilization of peri-islet tertiary lymphoid organs. Diabetes 2010, 59, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, C.; He, X.; Shang, W.; Bi, Y.; Wang, D. Long-term effect of FTY720 on lymphocyte count and islet allograft survival in mice. Microsurgery 2007, 27, 300–304. [Google Scholar] [CrossRef]
- Yin, N.; Zhang, N.; Xu, J.; Shi, Q.; Ding, Y.; Bromberg, J.S. Targeting lymphangiogenesis after islet transplantation prolongs islet allograft survival. Transplantation 2011, 92, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Cnop, M. Mining genes in type 2 diabetic islets and finding gold. Cell Metab. 2012, 16, 555–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Weir, G.C. Glucolipotoxicity, beta-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef]
- Oshima, M.; Pechberty, S.; Bellini, L.; Gopel, S.O.; Campana, M.; Rouch, C.; Dairou, J.; Cosentino, C.; Fantuzzi, F.; Toivonen, S.; et al. Stearoyl CoA desaturase is a gatekeeper that protects human beta cells against lipotoxicity and maintains their identity. Diabetologia 2020, 63, 395–409. [Google Scholar] [CrossRef] [Green Version]
- Gehrmann, W.; Wurdemann, W.; Plötz, T.; Jörns, A.; Lenzen, S.; Elsner, M. Antagonism Between Saturated and Unsaturated Fatty Acids in ROS Mediated Lipotoxicity in Rat Insulin-Producing Cells. Cell Physiol. Biochem. 2015, 36, 852–865. [Google Scholar] [CrossRef]
- Plötz, T.; Krümmel, B.; Laporte, A.; Pingitore, A.; Persaud, S.J.; Jörns, A.; Elsner, M.; Mehmeti, I.; Lenzen, S. The monounsaturated fatty acid oleate is the major physiological toxic free fatty acid for human beta cells. Nutr. Diabetes 2017, 7, 305. [Google Scholar] [CrossRef] [Green Version]
- Plötz, T.; von Hanstein, A.S.; Krümmel, B.; Laporte, A.; Mehmeti, I.; Lenzen, S. Structure-toxicity relationships of saturated and unsaturated free fatty acids for elucidating the lipotoxic effects in human EndoC-betaH1 beta-cells. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 165525. [Google Scholar] [CrossRef] [PubMed]
- Kelpe, C.L.; Moore, P.C.; Parazzoli, S.D.; Wicksteed, B.; Rhodes, C.J.; Poitout, V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J. Biol. Chem. 2003, 278, 30015–30021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Hanstein, A.S.; Lenzen, S.; Plötz, T. Toxicity of fatty acid profiles of popular edible oils in human EndoC-betaH1 beta-cells. Nutr. Diabetes 2020, 10, 5. [Google Scholar] [CrossRef]
- Janikiewicz, J.; Hanzelka, K.; Dziewulska, A.; Kozinski, K.; Dobrzyn, P.; Bernas, T.; Dobrzyn, A. Inhibition of SCD1 impairs palmitate-derived autophagy at the step of autophagosome-lysosome fusion in pancreatic beta-cells. J. Lipid Res. 2015, 56, 1901–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitton, J.; Bandet, C.L.; Mariko, M.L.; Tan-Chen, S.; Bourron, O.; Benomar, Y.; Hajduch, E.; Le Stunff, H. Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes. Cells 2020, 9, 1682. [Google Scholar] [CrossRef] [PubMed]
- Gjoni, E.; Brioschi, L.; Cinque, A.; Coant, N.; Islam, M.N.; Ng, C.K.; Verderio, C.; Magnan, C.; Riboni, L.; Viani, P.; et al. Glucolipotoxicity impairs ceramide flow from the endoplasmic reticulum to the Golgi apparatus in INS-1 beta-cells. PLoS ONE 2014, 9, e110875. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; MacIntosh, G.; Preston, A.M.; Bartley, C.; Busch, A.K.; Fuller, M.; Laybutt, D.R.; Meikle, P.J.; Biden, T.J. A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem. J. 2011, 435, 267–276. [Google Scholar] [CrossRef]
- Pearson, G.L.; Mellett, N.; Chu, K.Y.; Boslem, E.; Meikle, P.J.; Biden, T.J. A comprehensive lipidomic screen of pancreatic beta-cells using mass spectroscopy defines novel features of glucose-stimulated turnover of neutral lipids, sphingolipids and plasmalogens. Mol. Metab. 2016, 5, 404–414. [Google Scholar] [CrossRef]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, I.; Mastrandrea, L.D. Serum sphingolipids and inflammatory mediators in adolescents at risk for metabolic syndrome. Endocrine 2012, 41, 442–449. [Google Scholar] [CrossRef]
- Kowalski, G.M.; Carey, A.L.; Selathurai, A.; Kingwell, B.A.; Bruce, C.R. Plasma sphingosine-1-phosphate is elevated in obesity. PLoS ONE 2013, 8, e72449. [Google Scholar] [CrossRef] [Green Version]
- Raichur, S.; Brunner, B.; Bielohuby, M.; Hansen, G.; Pfenninger, A.; Wang, B.; Bruning, J.C.; Larsen, P.J.; Tennagels, N. The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Mol. Metab. 2019, 21, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Buschard, K.; Fredman, P.; Bog-Hansen, E.; Blomqvist, M.; Hedner, J.; Rastam, L.; Lindblad, U. Low serum concentration of sulfatide and presence of sulfated lactosylceramid are associated with Type 2 diabetes. The Skaraborg Project. Diabet. Med. A J. Br. Diabet. Assoc. 2005, 22, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Floegel, A.; Stefan, N.; Yu, Z.; Muhlenbruch, K.; Drogan, D.; Joost, H.G.; Fritsche, A.; Haring, H.U.; Hrabe de Angelis, M.; Peters, A.; et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 2013, 62, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Kanazawa, I.; Sugimoto, T. Visceral fat accumulation is associated with increased plasma sphingosine-1-phosphate levels in type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2018, 143, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Laychock, S.G.; Sessanna, S.M.; Lin, M.H.; Mastrandrea, L.D. Sphingosine 1-phosphate affects cytokine-induced apoptosis in rat pancreatic islet beta-cells. Endocrinology 2006, 147, 4705–4712. [Google Scholar] [CrossRef] [Green Version]
- Mastrandrea, L.D.; Sessanna, S.M.; Laychock, S.G. Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: Response to cytokines. Diabetes 2005, 54, 1429–1436. [Google Scholar] [CrossRef] [Green Version]
- Fyrst, H.; Saba, J.D. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat. Chem. Biol. 2010, 6, 489–497. [Google Scholar] [CrossRef]
- Toman, R.E.; Spiegel, S. Lysophospholipid receptors in the nervous system. Neurochem. Res. 2002, 27, 619–627. [Google Scholar] [CrossRef]
- Le Stunff, H.; Peterson, C.; Liu, H.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate and lipid phosphohydrolases. Biochim. Biophys. Acta 2002, 1582, 8–17. [Google Scholar] [CrossRef]
- Le Stunff, H.; Giussani, P.; Maceyka, M.; Lepine, S.; Milstien, S.; Spiegel, S. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 2007, 282, 34372–34380. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, A.; Saba, J.D. Truth and consequences of sphingosine-1-phosphate lyase. Adv. Biol. Regul. 2012, 52, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Sigal, Y.J.; McDermott, M.I.; Morris, A.J. Integral membrane lipid phosphatases/phosphotransferases: Common structure and diverse functions. Biochem. J. 2005, 387, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.D. Fifty years of lyase and a moment of truth: Sphingosine phosphate lyase from discovery to disease. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laychock, S.G.; Tian, Y.; Sessanna, S.M. Endothelial differentiation gene receptors in pancreatic islets and INS-1 cells. Diabetes 2003, 52, 1986–1993. [Google Scholar] [CrossRef] [Green Version]
- Kleuser, B. Divergent Role of Sphingosine 1-Phosphate in Liver Health and Disease. Int. J. Mol. Sci. 2018, 19, 1682. [Google Scholar] [CrossRef] [Green Version]
- Hagen, N.; Hans, M.; Hartmann, D.; Swandulla, D.; van Echten-Deckert, G. Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ. 2011, 18, 1356–1365. [Google Scholar] [CrossRef] [Green Version]
- Hagen, N.; Van Veldhoven, P.P.; Proia, R.L.; Park, H.; Merrill, A.H., Jr.; van Echten-Deckert, G. Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 2009, 284, 11346–11353. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, I.; van Echten-Deckert, G. Sphingosine 1-phosphate—A double edged sword in the brain. Biochim. Biophys. Acta. Biomembr. 2017, 1859, 1573–1582. [Google Scholar] [CrossRef]
- Mitroi, D.N.; Deutschmann, A.U.; Raucamp, M.; Karunakaran, I.; Glebov, K.; Hans, M.; Walter, J.; Saba, J.; Graler, M.; Ehninger, D.; et al. Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin- proteasome mediated mechanism. Sci. Rep. 2016, 6, 37064. [Google Scholar] [CrossRef] [PubMed]
- van Echten-Deckert, G.; Alam, S. Sphingolipid metabolism—An ambiguous regulator of autophagy in the brain. Biol. Chem. 2018, 399, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Holbling, B.V.; Schumak, B.; Hubner, M.P.; Graler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kon, J.; Tomura, H.; Osada, M.; Murata, N.; Kuwabara, A.; Watanabe, T.; Ohta, H.; Ui, M.; Okajima, F. Activation of phospholipase C-Ca2+ system by sphingosine 1-phosphate in CHO cells transfected with Edg-3, a putative lipid receptor. FEBS Lett. 1999, 443, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Ancellin, N.; Hla, T. Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J. Biol. Chem. 1999, 274, 18997–19002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonda, K.; Okamoto, H.; Takuwa, N.; Yatomi, Y.; Okazaki, H.; Sakurai, T.; Kimura, S.; Sillard, R.; Harii, K.; Takuwa, Y. The novel sphingosine 1-phosphate receptor AGR16 is coupled via pertussis toxin-sensitive and -insensitive G-proteins to multiple signalling pathways. Biochem. J. 1999, 337 Pt 1, 67–75. [Google Scholar] [CrossRef]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H., Jr.; Futerman, A.H. Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef] [Green Version]
- Panneer Selvam, S.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D.; et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 8, ra58. [Google Scholar] [CrossRef] [Green Version]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S.; Milstien, S.; Grant, S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 2012, 31, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Wang, W.; Li, N.; Yan, S.; Rong, K.; Lan, T.; Xia, P. Sphingosine kinase 2 promotes lipotoxicity in pancreatic beta-cells and the progression of diabetes. FASEB J. 2019, 33, 3636–3646. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floegel, A.; von Ruesten, A.; Drogan, D.; Schulze, M.B.; Prehn, C.; Adamski, J.; Pischon, T.; Boeing, H. Variation of serum metabolites related to habitual diet: A targeted metabolomic approach in EPIC-Potsdam. Eur. J. Clin. Nutr. 2013, 67, 1100–1108. [Google Scholar] [CrossRef]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Dettmer, R.; Niwolik, I.; Cirksena, K.; Yoshimoto, T.; Tang, Y.; Mehmeti, I.; Gurgul-Convey, E.; Naujok, O. Pro-inflammatory cytokines induce rapid, nitric oxygen-independent apoptosis, expression of chemotactic mediators and interleukin-32 secretion in human pluripotent stem cell-derived beta cells. Diabetologia, 2022; Online first February. [Google Scholar]
- Spiegel, S.; Milstien, S. Exogenous and intracellularly generated sphingosine 1-phosphate can regulate cellular processes by divergent pathways. Biochem. Soc. Trans. 2003, 31, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiu, V.; Leuti, A.; Maccarrone, M. Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Paradiso, E.; Lazzaretti, C.; Sperduti, S.; Antoniani, F.; Fornari, G.; Brigante, G.; Di Rocco, G.; Tagliavini, S.; Trenti, T.; Morini, D.; et al. Sphingosine-1 phosphate induces cAMP/PKA-independent phosphorylation of the cAMP response element-binding protein (CREB) in granulosa cells. Mol. Cell. Endocrinol. 2021, 520, 111082. [Google Scholar] [CrossRef]
- Means, C.K.; Brown, J.H. Sphingosine-1-phosphate receptor signalling in the heart. Cardiovasc. Res. 2009, 82, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Rutti, S.; Ehses, J.A.; Sibler, R.A.; Prazak, R.; Rohrer, L.; Georgopoulos, S.; Meier, D.T.; Niclauss, N.; Berney, T.; Donath, M.Y.; et al. Low—And high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology 2009, 150, 4521–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imasawa, T.; Koike, K.; Ishii, I.; Chun, J.; Yatomi, Y. Blockade of sphingosine 1-phosphate receptor 2 signaling attenuates streptozotocin-induced apoptosis of pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2010, 392, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirot, P.; Cardozo, A.K.; Eizirik, D.L. Mediators and mechanisms of pancreatic beta-cell death in type 1 diabetes. Arq Bras. Endocrinol. Metab. 2008, 52, 156–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Byun, H.S.; Bittman, R.; Saba, J.D. The sphingolipid degradation product trans-2-hexadecenal induces cytoskeletal reorganization and apoptosis in a JNK-dependent manner. Cell. Signal. 2011, 23, 1144–1152. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, P.; Kumar, A.; Byun, H.S.; Bittman, R.; Saba, J.D.; Hecht, S.S. The sphingolipid degradation product trans-2-hexadecenal forms adducts with DNA. Biochem. Biophys. Res. Commun. 2012, 424, 18–21. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Badeanlou, L.; Bielawski, J.; Ciaraldi, T.P.; Samad, F. Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E756–E768. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.M.; Chen, J.L.; Wang, G.G.; Wang, H.; Lu, Y.; Li, J.F.; Yi, J.; Yuan, Y.J.; Zhang, Q.W.; Mi, J.; et al. Sphingosine kinase 1 participates in insulin signalling and regulates glucose metabolism and homeostasis in KK/Ay diabetic mice. Diabetologia 2007, 50, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Kendall, M.R.; Hupfeld, C.J. FTY720, a sphingosine-1-phosphate receptor modulator, reverses high-fat diet-induced weight gain, insulin resistance and adipose tissue inflammation in C57BL/6 mice. Diabetes Obes. Metab. 2008, 10, 802–805. [Google Scholar]
- Mechtcheriakova, D.; Wlachos, A.; Sobanov, J.; Kopp, T.; Reuschel, R.; Bornancin, F.; Cai, R.; Zemann, B.; Urtz, N.; Stingl, G.; et al. Sphingosine 1-phosphate phosphatase 2 is induced during inflammatory responses. Cell. Signal. 2007, 19, 748–760. [Google Scholar] [CrossRef]
- Lepine, S.; Allegood, J.C.; Park, M.; Dent, P.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2011, 18, 350–361. [Google Scholar] [CrossRef]
- Alvarez, S.E.; Milstien, S.; Spiegel, S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. TEM 2007, 18, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Hla, T.; Lee, M.J.; Ancellin, N.; Paik, J.H.; Kluk, M.J. Lysophospholipids—Receptor revelations. Science 2001, 294, 1875–1878. [Google Scholar] [CrossRef] [PubMed]
- Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase in immunity and cancer: Silencing the siren. Trends Mol. Med. 2007, 13, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Saba, J.D. Lyase to live by: Sphingosine phosphate lyase as a therapeutic target. Expert Opin. Ther. Targets 2009, 13, 1013–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albi, E.; Alessenko, A.; Grosch, S. Sphingolipids in Inflammation. Mediat. Inflamm. 2018, 2018, 7464702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degagne, E.; Saba, J.D. S1pping fire: Sphingosine-1-phosphate signaling as an emerging target in inflammatory bowel disease and colitis-associated cancer. Clin. Exp. Gastroenterol. 2014, 7, 205–214. [Google Scholar]
- Degagne, E.; Pandurangan, A.; Bandhuvula, P.; Kumar, A.; Eltanawy, A.; Zhang, M.; Yoshinaga, Y.; Nefedov, M.; de Jong, P.J.; Fong, L.G.; et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J. Clin. Investig. 2014, 124, 5368–5384. [Google Scholar] [CrossRef] [Green Version]
- Snider, A.J.; Wu, B.X.; Jenkins, R.W.; Sticca, J.A.; Kawamori, T.; Hannun, Y.A.; Obeid, L.M. Loss of neutral ceramidase increases inflammation in a mouse model of inflammatory bowel disease. Prostaglandins Other Lipid Mediat. 2012, 99, 124–130. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Conditions | Effects | Ref. |
|---|---|---|
| Extracellular S1P alone | Nontoxic nM-5 µM < 24 h >5 µM inhibition of cell viability and GSIS >5 µM induction of Caspase-3 activation > 5µM inhibition of proliferation 5 µM weak induction of NFκB nM no induction of iNOS and NO Increase in cAMP No effect on ATP content Insulin secretion stimulation at 3 mM Glc Potentiation of GSIS (<5 µM) HDL-mediated inhibition of TUNEL staining after IL-1β or HG (mouse and human islets) S1P receptors specific (major role of S1P2 and S1P3) (MIN6, mouse islets; INS1, rat islets) | [25,26,30,105] [26,30] [26,30,105] [26,30] [30] [105] [30] [30] [30] [24,26,27,30] [141] [26,106,142] |
| Extracellular S1P and proinflammatory cytokines | Prevention of caspase-3 activation (INS1E) Decrease in TUNEL staining (INS1, rat islets) Inhibition of apoptosis (MIN6) Prevention of cytochrome c release (INS1) Prevention of viability loss (INS1E) Partial protection from proliferation inhibition (INS1E) No additive effect on NFκB (INS1E) No additive effect on iNOS and NO (INS1) S1P2 and S1P3-mediated effects (MIN6, mouse islets, INS1, rat islets) PKC-mediated effects (INS1) | [30,105] [105] [26] [105] [30] [30] [30] [105] [26,105] [142] [106,142] |
| Extracellular S1P and FFAs | Prevention of PA-mediated apoptosis (double staining with annexin V and propidium iodide, caspase-3 activation, TUNEL staining) (INS1, MIN6, mouse islets) Prevention of PA-mediated cell death (PI) (MIN6) | [26,28] [28] |
| Enzymes and S1P | Proinflammatory cytokines | Ref. | FFAs | Ref. |
|---|---|---|---|---|
| SK1 | Expression upregulated (1-8 h) Expression unaffected (24 h) Activity unaffected (1-24 h) Function not studied | [106] [30] [105,106] | Expression upregulated by PA (24 h) Activity upregulated by PA Overexpression protective against PA Suppression toxic against PA | [25] [26,27] [25] [25,28] |
| SK2 | Expression unaffected (1-8 h) Expression upregulated (24 h) Activity upregulated (15 min-8 h) Function not studied | [106] [30] [105,106] | Expression unaffected by PA (24 h) Expression upregulated by PA (24 h) Activity upregulated by PA? Overexpression deleterious against PA | [25] [132] [26,27] [132] |
| SPL | Expression mildly upregulated (6h) Expression downregulated (24 h) Overexpression protective (24 h) Suppression deleterious (24 h) | [30] [30] [30] [30] | Expression-no data Overexpression toxic against PA (24 h) Overexpression induces OA toxicity (24 h) Suppression protective in EndoC-βH1 cells (24 h) | [31] [31] [31] |
| S1P function | Based on SPL overexpression studies in INS1E cells S1P participates in early phase of cytokine toxicity (24 h): | Based on SK1 and SPL studies S1P protects against FFA toxicity; with exception of SK2-derived S1P that is deleterious (24 h): | ||
| Increase of S1P, but no release from cells Cell viability inhibition Caspase-3 activation Proliferation inhibition CHOP upregulation Calcium homeostasis disruption Mitochondrial chaperones down ATP content fall BAD dephosphorylation No effects on ROS generation GSIS inhibition | [106] [30] [30] [30] [30] [30] [30] [30] [30] [30] [30] | Increase of S1P and release from cells Cell viability Inhibition of apoptosis ER stress downregulation Inhibition of oxidative stress Prevention of ceramide generation Mitochondrial chaperones up Inhibition of cytochrome c release Lipid droplet formation GSIS potentiation | [25,26,105] [31] [25,28] [31] [31] [25] [31] [28] [31] [25,26,27,28,132] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurgul-Convey, E. To Be or Not to Be: The Divergent Action and Metabolism of Sphingosine-1 Phosphate in Pancreatic Beta-Cells in Response to Cytokines and Fatty Acids. Int. J. Mol. Sci. 2022, 23, 1638. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031638
Gurgul-Convey E. To Be or Not to Be: The Divergent Action and Metabolism of Sphingosine-1 Phosphate in Pancreatic Beta-Cells in Response to Cytokines and Fatty Acids. International Journal of Molecular Sciences. 2022; 23(3):1638. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031638
Chicago/Turabian StyleGurgul-Convey, Ewa. 2022. "To Be or Not to Be: The Divergent Action and Metabolism of Sphingosine-1 Phosphate in Pancreatic Beta-Cells in Response to Cytokines and Fatty Acids" International Journal of Molecular Sciences 23, no. 3: 1638. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031638