
COVID-19 and Lung Mast Cells: The Kallikrein–Kinin Activation Pathway

 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Results
3. Discussion
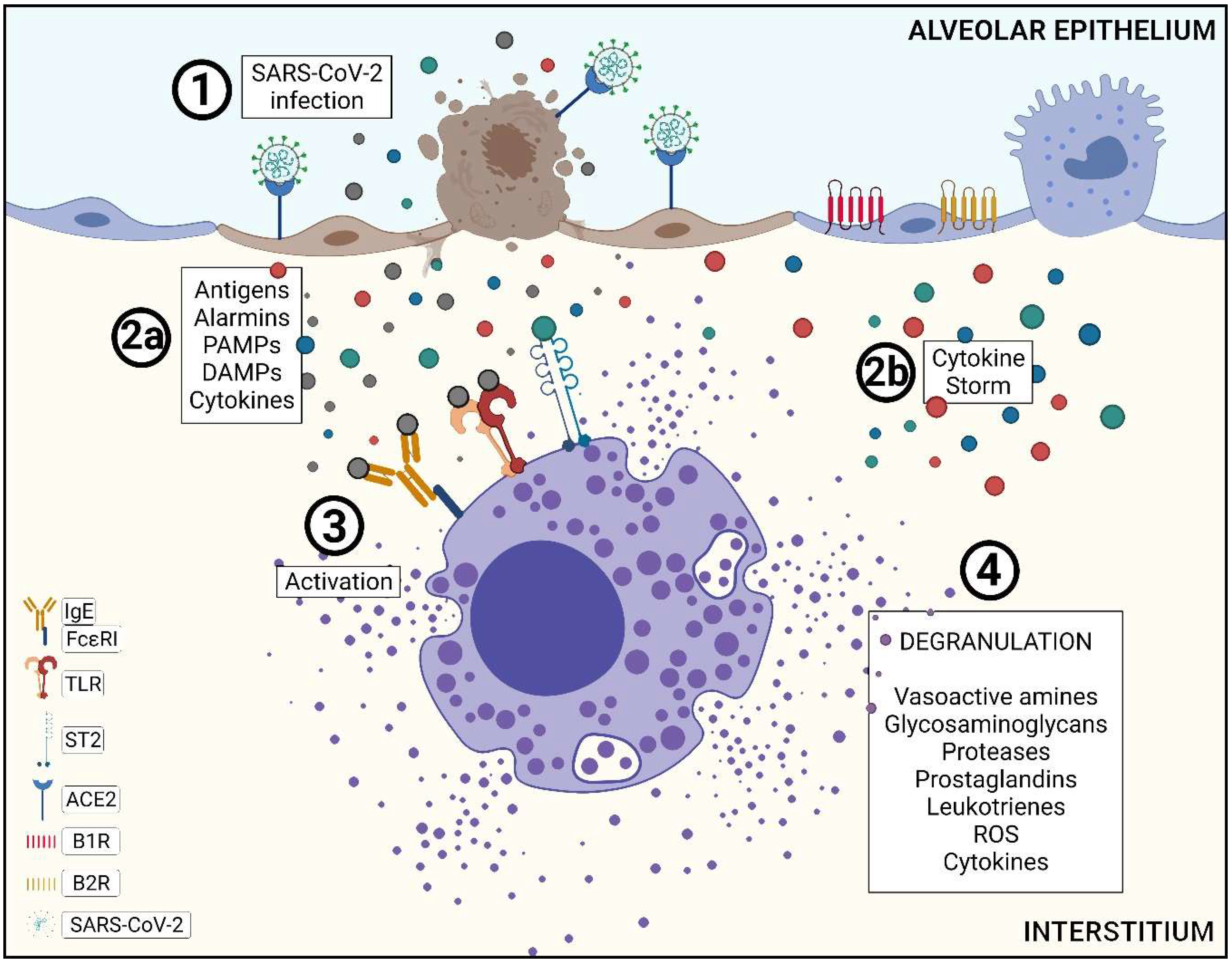
3.1. Pathophysiology of SARS-CoV-2 Infection in the Lower Airways and Mast Cells Activation
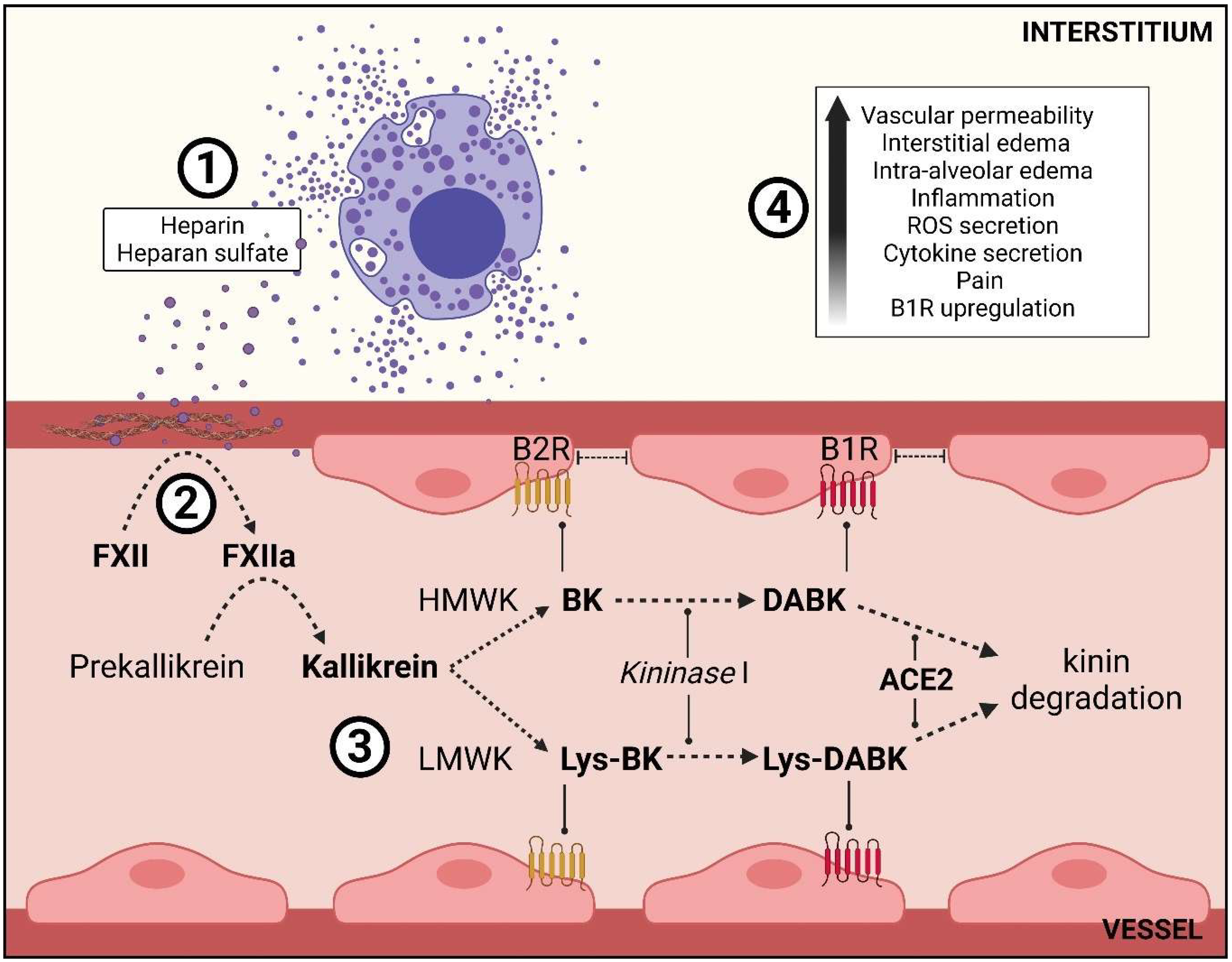
3.2. Mast Cell Degranulation and the Role of Its Mediators in the Kallikrein–Kinin System
4. Materials and Methods
4.1. Post-Mortem Samples and Histochemistry Assay
4.2. Immunohistochemistry Assay
4.3. Morphometric Analysis and MC-Counting Process
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://www.who.int/ (accessed on 20 January 2022).
- Simonsen, L.; Spreeuwenberg, P.; Lustig, R.; Taylor, R.J.; Fleming, D.M.; Kroneman, M.; Van Kerkhove, M.D.; Mounts, A.W.; Paget, W.J.; GLaMOR Collaborating Teams. Global Mortality Estimates for the 2009 Influenza Pandemic from the GLaMOR Project: A Modeling Study. PLoS Med. 2013, 10, 1001558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawood, F.S.; Iuliano, A.D.; Reed, C.; Meltzer, M.I.; Shay, D.K.; Cheng, P.Y.; Bandaranayake, D.; Breiman, R.F.; Brooks, W.A.; Buchy, P.; et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: A modelling study. Lancet Infect. Dis. 2012, 12, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gembardt, F.; Sterner-Kock, A.; Imboden, H.; Spalteholz, M.; Reibitz, F.; Schultheiss, H.P.; Siems, W.E.; Walther, T. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides 2005, 26, 1270–1277. [Google Scholar] [CrossRef]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Bader, M.; Ganten, D. Update on tissue renin-angiotensin systems. J. Mol. Med. 2008, 86, 615–621. [Google Scholar] [CrossRef]
- Miggiolaro, A.F.R.d.S.; Motta Junior, J.S.; de Paula, C.B.V.; Nagashima, S.; Malaquias, M.A.S.; Carstens, L.B.; Amaral, A.M.N.; Baena, C.P.; de Noronha, L. COVID-19 cytokine storm in pulmonary tissue: Anatomopathological and immunohistochemical findings. Diabetes Metab. Syndr. 2020, 14, 337–339. [Google Scholar]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Nagashima, S.; Mendes, M.C.; Camargo Martins, A.P.; Borges, N.H.; Godoy, T.M.; Miggiolaro, A.F.R.D.S.; da Silva Dezidério, F.; Machado-Souza, C.; De Noronha, L. Endothelial dysfunction and thrombosis in patients with COVID-19—Brief report. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2404–2407. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- González-Deolano, D.; Álvarez-Twose, I. Mast cells as key players in allergy and inflammation. J. Investig. Allergol. Clin. Immunol. 2018, 28, 365–378. [Google Scholar] [CrossRef]
- Amin, K. The role of mast cells in allergic inflammation. Respir. Med. 2012, 106, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef]
- Abraham, S.N.; John, A.L.S. Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 2010, 10, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Kunder, C.A.; St John, A.L.; Abraham, S.N. Mast cell modulation of the vascular and lymphatic endothelium. Blood 2011, 118, 5383–5393. [Google Scholar] [CrossRef]
- Schmaier, A.H. The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J. Thromb. Haemost. 2016, 14, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Oschatz, C.; Maas, C.; Lecher, B.; Jansen, T.; Björkqvist, J.; Tradler, T.; Sedlmeier, R.; Burfeind, P.; Cichon, S.; Hammerschmidt, S.; et al. Mast Cells Increase Vascular Permeability by Heparin-Initiated Bradykinin Formation In Vivo. Immunity 2011, 34, 258–268. [Google Scholar] [CrossRef]
- Ghebrehiwet, B.; Kaplan, A.P.; Joseph, K.; Peerschke, E.I.B. The complement and contact activation systems: Partnership in pathogenesis beyond angioedema. Immunol. Rev. 2016, 274, 281–289. [Google Scholar] [CrossRef]
- Sheikh, I.A.; Kaplan, A.P. Studies of the digestion of bradykinin, lysyl bradykinin, and kinin-degradation products by carboxypeptidases A, B, and N. Biochem. Pharmacol. 1986, 35, 1957–1963. [Google Scholar] [CrossRef]
- Sriramula, S. Kinin B1 receptor: A target for neuroinflammation in hypertension. Pharmacol. Res. 2020, 155, 104715. [Google Scholar] [CrossRef]
- Ji, B.; Shang, L.; Wang, C.; Wan, L.; Cheng, B.; Chen, J. Roles for heterodimerization of APJ and B2R in promoting cell proliferation via ERK1/2-eNOS signaling pathway. Cell Signal. 2020, 73, 109671. [Google Scholar] [CrossRef]
- Dagnino, A.P.A.; Campos, M.M.; Silva, R.B.M. Kinins and their receptors in infectious diseases. Pharmaceuticals 2020, 13, 215. [Google Scholar] [CrossRef]
- Marceau, F.; Lussier, A.; Regoli, D. Pharmacology of kinins: Their relevance to tissue injury and inflammation. Gen. Pharmac. 1983, 14, 209–229. [Google Scholar] [CrossRef]
- Adachi, T.; Chong, J.M.; Nakajima, N.; Sano, M.; Yamazaki, J.; Miyamoto, I.; Nishioka, H.; Akita, H.; Sato, Y.; Kataoka, M.; et al. Clinicopathologic and immunohistochemical findings from autopsy of patient with COVID-19, Japan. Emerg. Infect. Dis. 2020, 26, 2157–2161. [Google Scholar] [CrossRef]
- Hu, Y.; Jin, Y.; Han, D.; Zhang, G.; Cao, S.; Xie, J.; Xue, J.; Li, Y.; Meng, D.; Fan, X.; et al. Mast Cell-Induced Lung Injury in Mice Infected with H5N1 Influenza Virus. J. Virol. 2012, 86, 3347–3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, A.C.; Temple, R.M.; Obar, J.J. Mast cells and influenza A virus: Association with allergic responses and beyond. Front. Immunol. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Lai, Y.; Bernard, J.J.; MacLeod, D.T.; Cogen, A.L.; Moss, B.; Di Nardo, A. Skin mast cells protect mice against vaccinia virus by triggering mast cell receptor S1PR2 and releasing antimicrobial peptides. J. Immunol. 2012, 188, 345–357. Available online: https://pubmed.ncbi.nlm.nih.gov/22140255/ (accessed on 8 September 2021). [CrossRef] [PubMed] [Green Version]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 1564–1581. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, T.; Cai, D.; Hu, Z.; Liao, H.; Zhi, L.; Wei, H.; Zhang, Z.; Qiu, Y.; Wang, J.; et al. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. 2020, 53, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Motta Junior, J.D.S.; Miggiolaro, A.F.R.D.S.; Nagashima, S.; de Paula, C.B.V.; Baena, C.P.; Scharfstein, J.; de Noronha, L. Mast Cells in Alveolar Septa of COVID-19 Patients: A Pathogenic Pathway That May Link Interstitial Edema to Immunothrombosis. Front. Immunol. 2020, 11, 574862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ren, L.; Zhang, L.; Jin, Q.; Li, M.; Wang, J. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890. [Google Scholar] [CrossRef]
- Redegeld, F.A.; Yu, Y.; Kumari, S.; Charles, N.; Blank, U. Non-IgE mediated mast cell activation. Immunol Rev. 2018, 282, 87–113. [Google Scholar] [CrossRef]
- Graham, A.C.; Hilmer, K.M.; Zickovich, J.M.; Obar, J.J. Inflammatory Response of Mast Cells during Influenza A Virus Infection Is Mediated by Active Infection and RIG-I Signaling. J. Immunol. 2013, 190, 4676–4684. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.D.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui-Takeuchi, M.; Ushio, H.; Fukuda, M.; Yamada, T.; Niyonsaba, F.; Okumura, K.; Ogawa, H.; Ikeda, S. Roles of retinoic acid-inducible gene-I-like receptors (RLRs), Toll-like receptor (TLR) 3 and 2′-5′ oligoadenylate synthetase as viral recognition receptors on human mast cells in response to viral infection. Immunol. Res. 2015, 61, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Bhuyan, P.; Capodici, J.; Weissman, D. Small Interfering RNAs Mediate Sequence-Independent Gene Suppression and Induce Immune Activation by Signaling through Toll-Like Receptor 3. J. Immunol. 2004, 172, 6545–6549. [Google Scholar] [CrossRef] [Green Version]
- Mehraj, V.; Ponte, R.; Routy, J.P. The Dynamic Role of the IL-33/ST2 Axis in Chronic Viral-infections: Alarming and Adjuvanting the Immune Response. EBioMedicine 2016, 9, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Aoki, R.; Kawamura, T.; Goshima, F.; Ogawa, Y.; Nakae, S.; Nakao, A.; Moriishi, K.; Nishiyama, Y.; Shimada, S. Mast cells play a key role in host defense against herpes simplex virus infection through TNF-α and IL-6 production. J. Invest. Dermatol. 2013, 133, 2170–2179. [Google Scholar] [CrossRef] [Green Version]
- D’mello, A.X.P.; Sylvester, T.V.; Ramya, V.; Britto, F.P.; Shetty, P.K.; Jasphin, S. Metachromasia and Metachromatic Dyes: A review. Int. J. Adv. Health Sci. 2016, 2, 12–17. [Google Scholar]
- Atiakshin, D.; Samoilova, V.; Buchwalow, I.; Boecker, W.; Tiemann, M. Characterization of mast cell populations using different methods for their identification. Histochem. Cell Biol. 2017, 147, 683–694. [Google Scholar] [CrossRef]
- Atiakshin, D.A.; Shishkina, V.V.; Gerasimova, O.A.; Meshkova, V.Y.; Samodurova, N.Y.; Samoilenko, T.V.; Buchwalow, I.B.; Samoilova, V.E.; Tiemann, M. Combined histochemical approach in assessing tryptase expression in the mast cell population. Acta Histochem. 2021, 123, 151711. [Google Scholar] [CrossRef]
- Khoury, P.; Lyons, J.J. Mast cell activation in the context of elevated basal serum tryptase: Genetics and presentations. Curr. Allergy Asthma Rep. 2019, 19, 55. [Google Scholar] [CrossRef]
- Vitte, J. Human mast cell tryptase in biology and medicine. Mol. Immunol. 2015, 63, 18–24. [Google Scholar] [CrossRef]
- Maun, H.R.; Jackman, J.K.; Choy, D.F.; Loyet, K.M.; Staton, T.L.; Jia, G.; Dressen, A.; Hackney, J.A.; Bremer, M.; Walters, B.T.; et al. An Allosteric Anti-tryptase Antibody for the Treatment of Mast Cell-Mediated Severe Asthma. Cell 2019, 179, 417–431.e19. [Google Scholar] [CrossRef] [Green Version]
- Heutinck, K.M.; ten Berge, I.J.M.; Hack, C.E.; Hamann, J.; Rowshani, A.T. Serine proteases of the human immune system in health and disease. Mol. Immunol. 2010, 47, 1943–1955. [Google Scholar] [CrossRef]
- Payne, V.; Kam, P.C.A. Mast cell tryptase: A review of its physiology and clinical significance. Anaesthesia 2004, 59, 695–703. [Google Scholar] [CrossRef]
- Caughey, G.H. Mast cell tryptases and chymases in inflammation and host defense. Immunol. Rev. 2007, 71, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, J.; Lindholt, J.S.; Sukhova, G.K.; Sinnamon, M.; Stevens, R.L.; Adachi, R.; Libby, P.; Thompson, R.W.; Shi, G.P. Mast cell tryptase deficiency attenuates mouse abdominal aortic aneurysm formation. Circ. Res. 2011, 108, 1316–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharfstein, J. Subverting bradykinin-evoked inflammation by co-opting the contact system: Lessons from survival strategies of Trypanosoma cruzi. Curr. Opin. Hematol. 2018, 25, 347–357. [Google Scholar] [CrossRef]
- Naudin, C.; Burillo, E.; Blankenberg, S.; Butler, L.; Renné, T. Factor XII Contact Activation. Semin. Thromb. Hemost. 2017, 43, 814–826. [Google Scholar] [CrossRef]
- Woywodt, A.; Bahlmann, F.H.; De Groot, K.; Haller, H.; Haubitz, M. Circulating endothelial cells: Life, death, detachment and repair of the endothelial cell layer. Nephrol. Dial. Transplant. 2002, 17, 1728–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivas, D.; Roldán, V.; Esteve-Pastor, M.A.; Roldán, I.; Tello-Montoliu, A.; Ruiz-Nodar, J.M.; Cosín-Sales, J.; Gámez, J.M.; Consuegra, L.; Ferreiro, J.L.; et al. Recomendaciones sobre el tratamiento antitrombótico durante la pandemia COVID-19. Posicionamiento del Grupo de Trabajo de Trombosis Cardiovascular de la Sociedad Española de Cardiología. Rev. Esp. Cardiol. 2020, 73, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Renné, T. The procoagulant and proinflammatory plasma contact system. Semin. Immunopathol. 2012, 34, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Sala-Cunill, A.; Björkqvist, J.; Senter, R.; Guilarte, M.; Cardona, V.; Labrador, M.; Nickel, K.F.; Butler, L.; Luengo, O.; Kumar, P.; et al. Plasma contact system activation drives anaphylaxis in severe mast cell-mediated allergic reactions. J. Allergy Clin. Immunol. 2015, 135, 1031–1043.e6. [Google Scholar] [CrossRef]
- Lalmanach, G.; Naudin, C.; Lecaille, F.; Fritz, H. Kininogens: More than cysteine protease inhibitors and kinin precursors. Biochimie 2010, 92, 1568–1579. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, Epidemiology, Pathogenesis, and Control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, P.R.; Sirois, P.; Fernandes, P.D. The role of kallikrein-kinin and renin-angiotensin systems in COVID-19 infection. Peptides 2021, 135, 170428. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray Jr, P.B.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Kuhr, F.; Lowry, J.; Zhang, Y.; Brovkovych, V.; Skidgel, R.A. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 2010, 44, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Sangsree, S.; Brovkovych, V.; Minshall, R.D.; Skidgel, R.A. Kininase I-type carboxypeptidases enhance nitric oxide production in endothelial cells by generating bradykinin B1 receptor agonists. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, 1959–1968. [Google Scholar] [CrossRef]
- Chen, Y.; Li, L. SARS-CoV-2: Virus dynamics and host response. Lancet Infect. Dis. 2020, 20, 515–516. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.A.; Kwok, S.; Berry, G.J.; Montine, T.J. Angiotensin-converting enzyme 2 (ACE2) expression increases with age in patients requiring mechanical ventilation. PLoS ONE 2021, 16, e0247060. [Google Scholar]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Nicolau, L.A.D.; Magalhães, P.J.C.; Vale, M.L. What would Sérgio Ferreira say to your physician in this war against COVID-19: How about kallikrein/kinin system? Med. Hypotheses 2020, 143, 109886. [Google Scholar] [CrossRef]
- Alhenc-Gelas, F.; Drueke, T.B. Blockade of SARS-CoV-2 infection by recombinant soluble ACE2. Kidney Int. 2020, 97, 1091–1093. [Google Scholar] [CrossRef]
- Batlle, D.; Wysocki, J.; Satchell, K. Soluble angiotensin-converting enzyme 2: A potential approach for coronavirus infection therapy? Clin. Sci. 2020, 134, 543–545. [Google Scholar] [CrossRef] [Green Version]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a ras-mediated bradykinin storm. eLife 2020, 9, 1–16. [Google Scholar] [CrossRef]
- Mori, T.; Abe, N.; Saito, K.; Toyama, H.; Endo, Y.; Ejima, Y.; Yamauchi, M.; Goto, M.; Mushiake, H.; Kazama, I. Hydrocortisone and dexamethasone dose-dependently stabilize mast cells derived from rat peritoneum. Pharmacol. Rep. 2016, 68, 1358–1365. [Google Scholar] [CrossRef]
- de Frias Carvalho, V.; de Oliveira Barreto, E.; Farias-Filho, F.A.; Gomes, L.H.F.; de Lima Mendonça, L.; Cordeiro, R.S.B.; Martins, M.A.; Silva, P.M.R.E. Reduced expression of IL-3 mediates intestinal mast cell depletion in diabetic rats: Role of insulin and glucocorticoid hormones. Int. J. Exp. Pathol. 2009, 90, 148–155. [Google Scholar] [CrossRef]
- Mekori, Y.A.; Oh, C.K.; Metcalfe, D.D. IL-3-dependent murine mast cells undergo apoptosis on removal of IL-3. Prevention of apoptosis by c-kit ligand. J. Immunol. 1993, 151, 3775–3784. [Google Scholar]
- Oray, M.; Abu Samra, K.; Ebrahimiadib, N.; Meese, H.; Foster, C.S. Long-term side effects of glucocorticoids. Expert Opin. Drug Saf. 2016, 15, 457–465. [Google Scholar] [CrossRef]
- Adcock, I.M.; Mumby, S. Glucocorticoids; Handbook of Experimental Pharmacology; Springer: New York, NY, USA, 2016; Volume 237, pp. 171–196. Available online: https://0-link-springer-com.brum.beds.ac.uk/chapter/10.1007/164_2016_98. (accessed on 19 September 2021).
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [PubMed]
- NIH—National Institute of Health. COVID-19 Treatment Guidelines: Corticosteroids. Available online: https://www.covid19treatmentguidelines.nih.gov/therapies/immunomodulators/corticosteroids/ (accessed on 6 September 2021).
- NIH—National Institute of Health. Hospitalized Adults: Therapeutic Management. COVID-19 Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/management/clinical-management/hospitalized-adults--therapeutic-management/ (accessed on 7 September 2021).
- Weng, Z.; Patel, A.B.; Panagiotidou, S.; Theoharides, T.C. The novel flavone tetramethoxyluteolin is a potent inhibitor of human mast cells. J. Allergy Clin. Immunol. 2014, 135, 1044–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theoharides, T.C. COVID-19, pulmonary mast cells, cytokine storms, and beneficial actions of luteolin. Biofactors 2020, 46, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Yu, B.; Zhang, W.; Zhang, W.; Xiao, Z.; Mao, Z.; Lai, Y.; Lin, D.; Ma, Q.; Pan, E.; et al. Toll-like receptor 2-mediated MAPKs and NF-κB activation requires the GNAO1-dependent pathway in human mast cells. Integr. Biol. 2016, 8, 968–975. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | H1N1 | COVID-19 | Control | |||
|---|---|---|---|---|---|---|
| Values | p-Value 1 | Values | p-Value 2 | Values | ||
| Gender a. | Male | 8 (80%) | 0.437 ** | 15 (62.5%) | 0.709 ** | 8 (72.7%) |
| Female | 2 (20%) | 9 (37.5%) | 3 (27.3%) | |||
| Age b | 43.5 ± 14 | 0.001 *** | 72 ± 12.5 | 0.001 *** | 42.3 ± 14.3 | |
| Time from HospitalizationTo Death b | 4.7 ± 6.13 | 0.003 *** | 15.9 ± 10.2 | 0.051 *** | 7.64 ± 13.1 | |
| Time of MechanicalVentilation b | 4.7 ± 6.13 | 0.028 *** | 12 ± 9.20 | - | - | |
| Alveolar Edema a | Present | 9 (90%) | 0.644 ** | 19 (79.2%) | 0.022 ** | 4 (36.4%) |
| Absent | 1 (10%) | 5 (20.8%) | 7 (63.6%) | |||
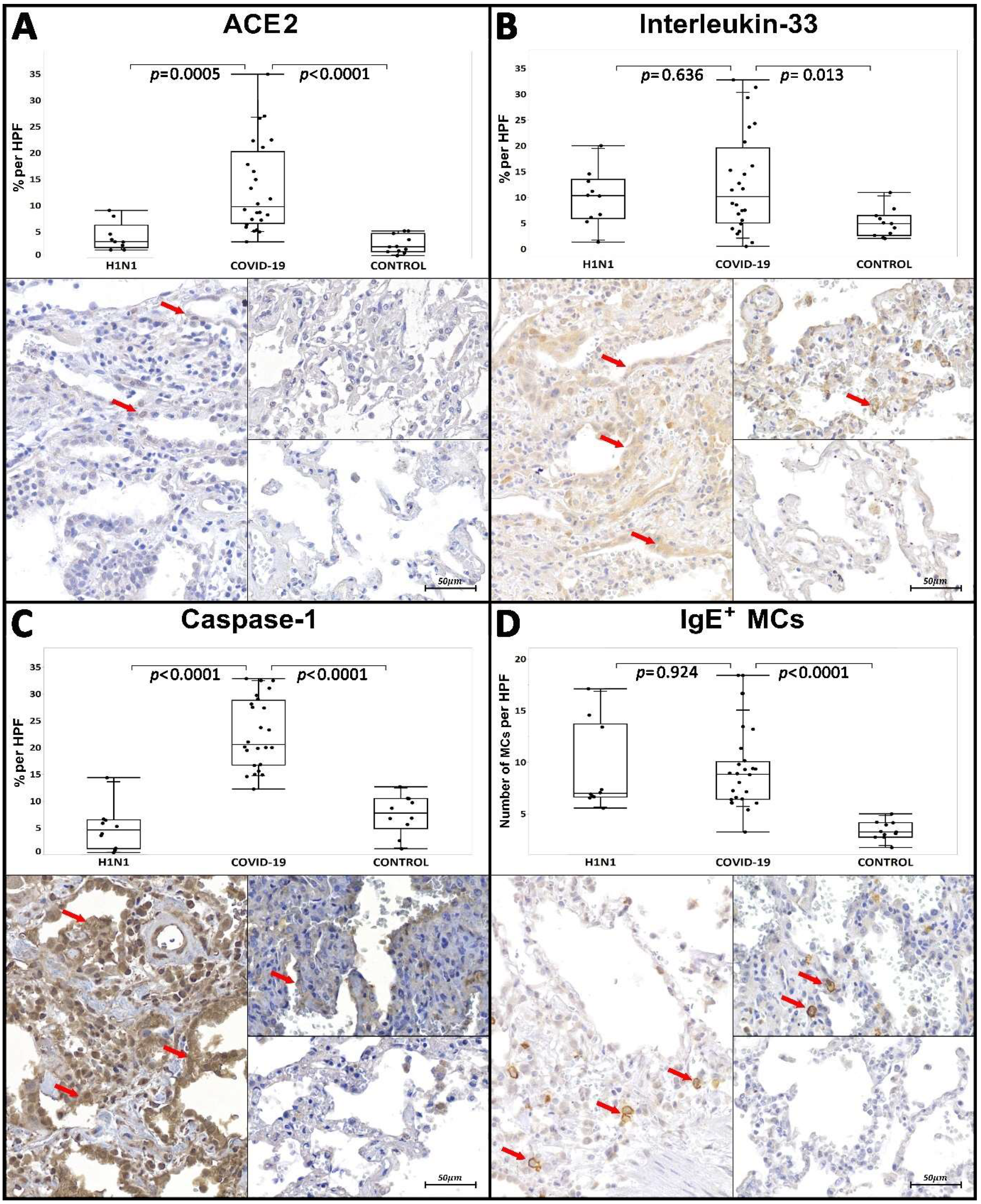
| Tissue Immunoexpression of ACE2 c | 3.07/4.26 (1.51–9.03) | 0.0005 * | 9.77/13.66 (3.1–34.95) | <0.0001 * | 2.14/3.46 (0.46–5.16) | |
| Tissue Immunoexpression of IL-33 c | 10.34/7.51 (1.36–19.45) | 0.636 * | 10.14/14.51 (0.52–32.75) | 0.013 * | 4.93/3.82 (2.05–10.94) | |
| Tissue Immunoexpression of CASP-1 c | 4.57/5,37 (0.36–14.34) | <0.0001 * | 20.56/12.1 (12.23–32.85) | <0.0001 * | 7.73/5.61 (1.09–12.67) | |
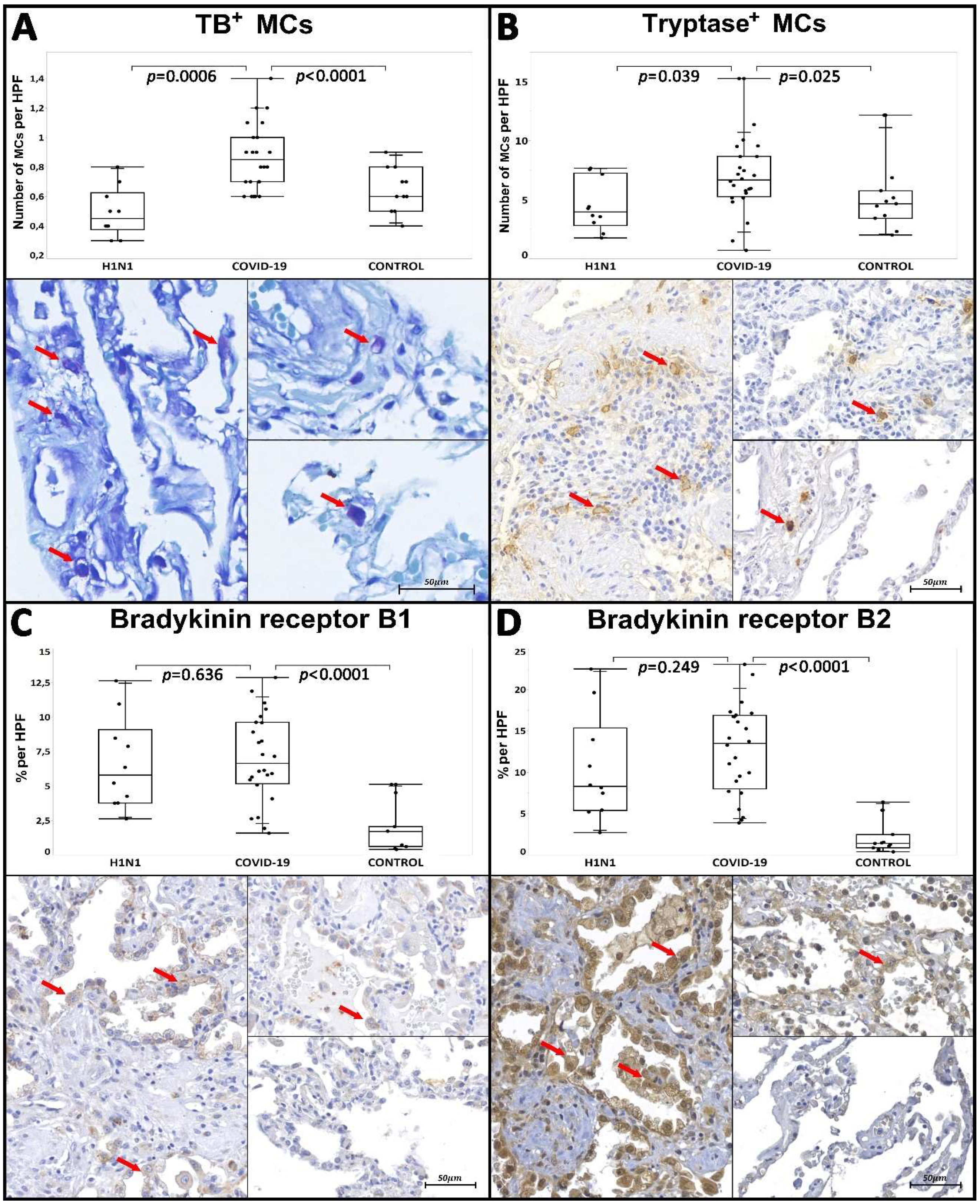
| Tissue Immunoexpression of B1R c | 5.79/7.34 (2.62–12.64) | 0.636 * | 6.65/4.45 (1.58–12.88) | <0.0001 * | 1.69/1.65 (0.39–5.11) | |
| Tissue Immunoexpression of B2R c | 8.29/10 (2.71–22.49) | 0.249 * | 13.51/8.87 (3.88–23.06) | <0.0001 * | 1.39/1.62 (0.38–6.4) | |
| Number of IgE+ MCs d | 6.97/7.06 (5.55–17.1) | 0.924 * | 8.85/3.65 (3.25–18.4) | <0.0001 * | 3.25/1.4 (1.75–5) | |
| Number of Tryptase+ MCs d | 3.95/4.44 (1.75–7.65) | 0.039 * | 6.65/3.4 (0.7–15.25) | 0.025 * | 4.65/2.33 (2–12.15) | |
| Number of TB Metachromatic MCs d | 0.45/0.25 (0.3–0.8) | 0.0006 * | 0.85/0.3 (0.6–1.4) | <0.0001 * | 0.6/0.3 (0.4–0.9) | |
| Characteristics | Number of Tryptase+ Mcs 2 | Number of Toluidine Blue Metachromatic Mcs 2 | ||
|---|---|---|---|---|
| Values | p-Value a | Values | p-Value b | |
| Treated 17/24 (70.8%) 1 | 6.2/2.7 (3–11.35) | 0.633 * | 1.2/0.77 (0.35–2.45) | 0.774 * |
| Non-Treated 7/24 (29.1%) 1 | 7.7/8.05 (0.7–15.25) | 1/1.3 (0.55–2.75) | ||
| Corticosterids Administered 3,4 | Dexamethasone 6 mg/day 12/17 Hydrocortisone 100 mg/day 1/17 Hydrocortisone 200 mg/day 3/17 Methylprednisolone 125 mg/day 2/17 Prednisone 60 mg/day 1/17 Prednisone 10 mg/day 1/17 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagashima, S.; Dutra, A.A.; Arantes, M.P.; Zeni, R.C.; Klein, C.K.; de Oliveira, F.C.; Piper, G.W.; Brenny, I.D.; Pereira, M.R.C.; Stocco, R.B.; et al. COVID-19 and Lung Mast Cells: The Kallikrein–Kinin Activation Pathway. Int. J. Mol. Sci. 2022, 23, 1714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031714
Nagashima S, Dutra AA, Arantes MP, Zeni RC, Klein CK, de Oliveira FC, Piper GW, Brenny ID, Pereira MRC, Stocco RB, et al. COVID-19 and Lung Mast Cells: The Kallikrein–Kinin Activation Pathway. International Journal of Molecular Sciences. 2022; 23(3):1714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031714
Chicago/Turabian StyleNagashima, Seigo, Anderson Azevedo Dutra, Mayara Pezzini Arantes, Rafaela Chiuco Zeni, Carolline Konzen Klein, Flávia Centenaro de Oliveira, Giulia Werner Piper, Isadora Drews Brenny, Marcos Roberto Curcio Pereira, Rebecca Benicio Stocco, and et al. 2022. "COVID-19 and Lung Mast Cells: The Kallikrein–Kinin Activation Pathway" International Journal of Molecular Sciences 23, no. 3: 1714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031714