
Hippocampal Over-Expression of Cyclooxygenase-2 (COX-2) Is Associated with Susceptibility to Stress-Induced Anhedonia in Mice


 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
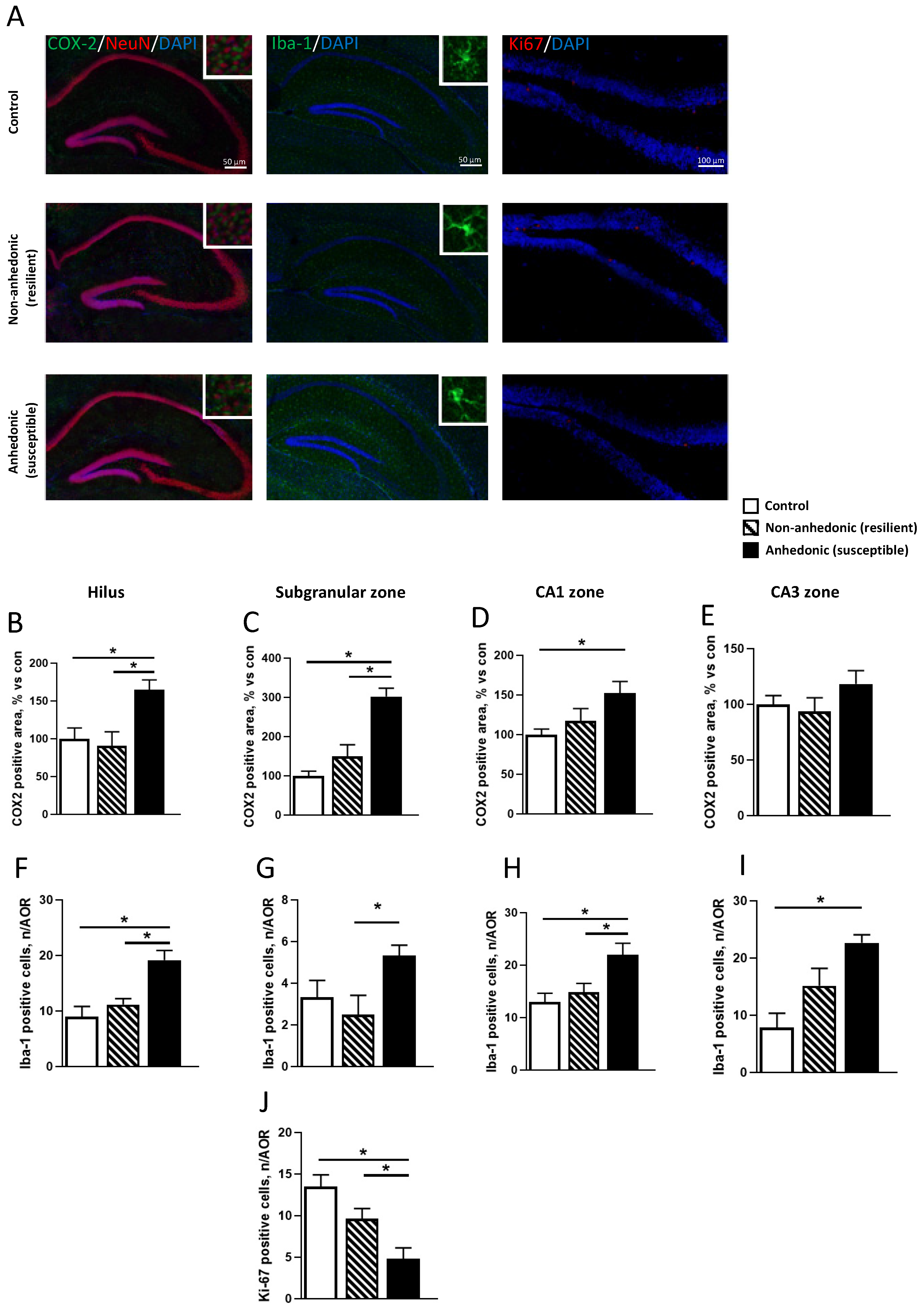
2.1. Expression of COX-2 in the Hippocampi of CMS Mice
2.2. Immunohistochemistry for COX-2, Iba-1, and Ki67 Expression in the Hippocampi of Mice Resilient and Susceptible to CMS-Induced Anhedonia
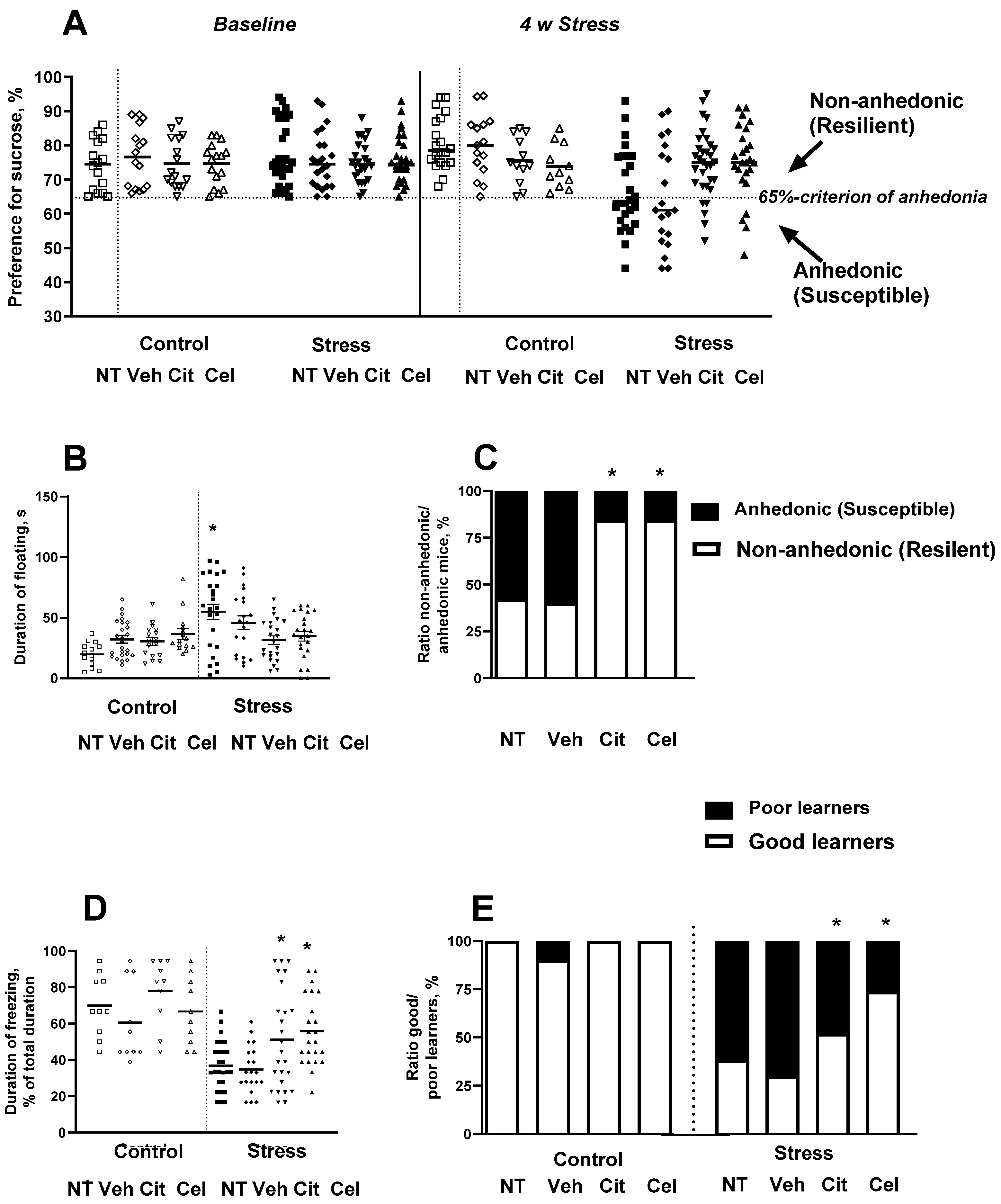
2.3. Effects of Chronic Treatment with Celecoxib and Citalopram on the Development of Stress-Induced Anhedonia and Depressive-like Syndrome
2.4. Acute Administration of Celecoxib Reduces Floating in the Porsolt Test
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Chronic Stress Experiments
4.3. A Study with the Porsolt Test
4.4. Chronic Stress Procedure and Determination of Anhedonia
4.5. Sucrose Preference Test
4.6. Forced Swim Test
4.7. Fear Conditioning Paradigm
4.8. A Two-Day Forced Swimming Porsolt Test and Drug Administration
4.9. Administration of Drugs
4.10. Culling and Brain Dissection
4.11. RNA Extraction and RT-PCR
4.12. Immunohistochemical Analysis of COX-2-Positive Cells in the Brain
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dale, E.; Bang-Andersen, B.; Sánchez, C. Emerging mechanisms and treatments for depression beyond SSRIs and SNRIs. Biochem. Pharmacol. 2015, 95, 81–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasin, D.S.; Sarvet, A.L.; Meyers, J.L.; Saha, T.D.; Ruan, W.J.; Stohl, M.; Grant, B.F. Epidemiology of Adult DSM-5 Major Depressive Disorder and Its Specifiers in the United States. JAMA Psychiatry 2018, 75, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, G.; Mucha, L.; Shi, S.; Guerin, A. Economic burden of relapse/recurrence in patients with major depressive disorder. J. Drug Assess. 2019, 8, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. World Federation for Mental Health. Depression: A Global Crisis; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Chaturvedi, S.K. COVID-19, Coronavirus and Mental Health Rehabilitation at Times of Crisis. J. Psychosoc. Rehabil. Ment. Health 2020, 2, 1–2. [Google Scholar] [CrossRef]
- Wind, T.R.; Rijkeboer, M.; Andersson, G.; Riper, H. The COVID-19 pandemic: The ‘black swan’ for mental health care and a turning point for e-health. Internet Interv. 2020, 20, 100317. [Google Scholar] [CrossRef]
- Alonso, J.; Vilagut, G.; Mortier, P.; Ferrer, M.; Alayo, I.; Aragón-Peña, A.; Aragonès, E.; Campos, M.; Cura-González, I.D.; Emparanza, J.I.; et al. Mental health impact of the first wave of COVID-19 pandemic on Spanish healthcare workers: A large cross-sectional survey. Rev. Psiquiatr. Salud Ment. 2021, 14, 90–105. [Google Scholar] [CrossRef]
- World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Zuzarte, P.; Duong, A.; Figueira, M.L.; Costa-Vitali, A.; Scola, G. Current Therapeutic Approaches for Targeting Inflammation in Depression and Cardiovascular Disease. Curr. Drug Metab. 2018, 19, 674–687. [Google Scholar] [CrossRef]
- Safer, D.J.; Zito, J.M. Short- and Long-Term Antidepressant Clinical Trials for Major Depressive Disorder in Youth: Findings and Concerns. Front. Psychiatry 2019, 10, 705. [Google Scholar] [CrossRef]
- Goh, K.K.; Chang, S.C.; Chen, C.H.; Lu, M.L. Therapeutic Strategies for Treatment-resistant Depression: State of the Art and Future Perspectives. Curr. Pharm. Des. 2020, 26, 244–252. [Google Scholar] [CrossRef]
- Orsolini, L.; Bellantuono, C. Serotonin reuptake inhibitors and breastfeeding: A systematic review. Hum. Psychopharmacol. Clin. Exp. 2015, 30, 4–20. [Google Scholar] [CrossRef]
- Quagliato, L.A.; Cosci, F.; Shader, R.I.; Silberman, E.K.; Starcevic, V.; Balon, R.; Dubovsky, S.L.; Salzman, C.; Krystal, J.H.; Weintraub, S.J.; et al. Selective serotonin reuptake inhibitors and benzodiazepines in panic disorder: A metaanalysis of common side effects in acute treatment. J. Psychopharmacol. 2019, 33, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Emmerzaal, T.L.; Nijkamp, G.; Veldic, M.; Rahman, S.; Andreazza, A.C.; Morava, E.; Rodenburg, R.J.; Kozicz, T. Effect of neuropsychiatric medications on mitochondrial function: For better or for worse. Neurosci. Biobehav. Rev. 2021, 127, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, S.L. What Is New about New Antidepressants. Psychother. Psychosom. 2018, 87, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Demin, K.A.; Sysoev, M.; Chernysh, M.V.; Savva, A.K.; Koshiba, M.; Wappler-Guzzetta, E.A.; Song, C.; De Abreu, M.S.; Leonard, B.; Parker, M.O.; et al. Animal models of major depressive disorder and the implications for drug discovery and development. Expert Opin. Drug Discov. 2019, 14, 365–378. [Google Scholar] [CrossRef]
- Mulinari, S. Monoamine theories of depression: Historical impact on biomedical research. J. Hist. Neurosci. 2012, 21, 366–392. [Google Scholar] [CrossRef]
- Maffioletti, E.; Minelli, A.; Tardito, D.; Gennarelli, M. Blues in the Brain and Beyond: Molecular Bases of Major Depressive Disorder and Relative Pharmacological and Non-Pharmacological Treatments. Genes 2020, 11, 1089. [Google Scholar] [CrossRef]
- Wegener, G.; Mathe, A.A.; Neumann, I.D. Selectively bred rodents as models of depression and anxiety. Curr. Top. Behav. Neurosci. 2012, 12, 139–187. [Google Scholar]
- Harro, J. Animal models of depression vulnerability. Curr. Top. Behav. Neurosci. 2013, 14, 29–54. [Google Scholar]
- Harro, J. Animal models of depression: Pros and cons. Cell Tissue Res. 2019, 377, 5–20. [Google Scholar] [CrossRef]
- Strekalova, T.; Liu, Y.; Kiselev, D.; Khairuddin, S.; Chiu, J.L.Y.; Lam, J.; Chan, Y.S.; Pavlov, D.; Proshin, A.; Lesch, K.P.; et al. Chronic mild stress paradigm as a rat model of depression: Facts, artifacts, and future perspectives. Psychopharmacology 2022, 24, 1–31. [Google Scholar] [CrossRef]
- Maes, M.; Meltzer, H.Y.; Bosmans, E.; Bergmans, R.; Vandoolaeghe, E.; Ranjan, R.; Desnyder, R. Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J. Affect. Disord. 1995, 34, 301–309. [Google Scholar] [CrossRef]
- Dantzer, R. Cytokine, sickness behavior, and depression. Immunol. Allergy Clin. N. Am. 2009, 29, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisman, H.; Merali, Z. Cytokines, stress, and depressive illness. Brain Behav. Immun. 2002, 16, 513–524. [Google Scholar] [CrossRef]
- Kopschina Feltes, P.; Doorduin, J.; Klein, H.C.; Juárez-Orozco, L.E.; Dierckx, R.A.; Moriguchi-Jeckel, C.M.; de Vries, E.F. Anti-inflammatory treatment for major depressive disorder: Implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J. Psychopharmacol. 2017, 31, 1149–1165. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.H.; Yin, L.Q.; Guan, L.P.; Li, Z.H.; Tan, C. Screening of chalcone analogs with anti-depressant, anti-inflammatory, analgesic, and COX-2-inhibiting effects. Bioorg. Med. Chem. Lett. 2020, 30, 127173. [Google Scholar] [CrossRef]
- Inyang, K.E.; Folger, J.K.; Laumet, G. Can FDA-Approved Immunomodulatory Drugs be Repurposed/Repositioned to Alleviate Chronic Pain? J. Neuroimmune Pharmacol. 2021, 16, 531–547. [Google Scholar] [CrossRef]
- Xie, W.L.; Chipman, J.G.; Robertson, D.L.; Erikson, R.L.; Simmons, D.L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl. Acad. Sci. USA 1991, 88, 2692–2696. [Google Scholar] [CrossRef] [Green Version]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Qiu, J.; Li, Q.; Bell, K.A.; Du, Y.; Jung, D.W.; Lee, J.Y.; Hao, J.; Jiang, J. Cyclooxygenase-2 contributes to oxidopamine-mediated neuronal inflammation and injury via the prostaglandin E2 receptor EP2 subtype. Sci. Rep. 2017, 7, 9459. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K.; Andreasson, K.I.; Kaufmann, W.E.; Barnes, C.A.; Worley, P.F. Expression of a mitogen-inducible cyclooxygenase in brain neurons: Regulation by synaptic activity and glucocorticoids. Neuron 1993, 11, 371–386. [Google Scholar] [CrossRef]
- Hoffman, C. COX-2 in brain and spinal cord implications for therapeutic use. Curr. Med. Chem. 2000, 7, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Young, J.L.; Jazaeri, A.A.; Darus, C.J.; Modesitt, S.C. Cyclooxygenase-2 in cervical neoplasia: A review. Gynecol. Oncol. 2008, 109, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.E.; Worley, P.F.; Pegg, J.; Bremer, M.; Isakson, P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc. Natl. Acad. Sci. USA 1996, 93, 2317–2321. [Google Scholar] [CrossRef] [Green Version]
- Breder, C.D.; Dewitt, D.; Kraig, R.P. Characterization of inducible cyclooxygenase in rat brain. J. Comp. Neurol. 1995, 355, 296–315. [Google Scholar] [CrossRef]
- Song, Q.; Fan, C.; Wang, P.; Li, Y.; Yang, M.; Yu, S.Y. Hippocampal CA1 βCaMKII mediates neuroinflammatory responses via COX-2/PGE2 signaling pathways in depression. J. Neuroinflamm. 2018, 15, 338. [Google Scholar] [CrossRef] [Green Version]
- Prabhakaran, J.; Molotkov, A.; Mintz, A.; Mann, J.J. Progress in PET Imaging of Neuroinflammation Targeting COX-2 Enzyme. Molecules 2021, 26, 3208. [Google Scholar] [CrossRef]
- Nakayama, M.; Uchimura, K.; Zhu, R.L.; Nagayama, T.; Rose, M.E.; Stetler, R.A.; Isakson, P.C.; Chen, J.; Graham, S.H. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc. Natl. Acad. Sci. USA 1998, 95, 10954–10959. [Google Scholar] [CrossRef] [Green Version]
- Koistinaho, J.; Koponen, S.; Chan, P.H. Expression of cyclooxygenase-2 mRNA after global ischemia is regulated by AMPA receptors and glucocorticoids. Stroke 1999, 30, 1900–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C. The interaction between cytokines and neurotransmitters in depression and stress: Possible mechanism of antidepressant treatments. Hum. Psychopharmacol. 2000, 15, 199–211. [Google Scholar] [CrossRef]
- Engblom, D.; Ek, M.; Saha, S.; Ericsson-Dahlstrand, A.; Jakobsson, P.J.; Blomqvist, A. Prostaglandins as inflammatory messengers across the blood-brain barrier. J. Mol. Med. 2002, 80, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Li, X.; Kang, Z.; Kadotomi, Y. Omega-3 fatty acid ethyl-eicosapentaenoate attenuates IL-1beta-induced changes in dopamine and metabolites in the shell of the nucleus accumbens: Involved with PLA2 activity and corticosterone secretion. Neuropsychopharmacology 2007, 32, 736–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.; Rahman, A.; Hasnain, A.; Lalonde, M.; Goldberg, V.M.; Haqqi, T.M. Green tea polyphenol epigallocatechin-3-gallate inhibits the IL-1 beta-induced activity and expression of cyclooxygenase-2 and nitric oxide synthase-2 in human chondrocytes. Free Radic. Biol. Med. 2002, 33, 1097–1105. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Ichikawa, Y.; Igarashi, O.; Ikeda, K.; Konno, S.; Aoyagi, J.; Kinoshita, M. Temocapril prevents motor neuron damage and upregulation of cyclooxygenase-II in glutamate-induced neurotoxicity. Neurol. Res. 2003, 25, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, S.; Hori, T.; Suzuki, T.; Baba, A.; Shiraishi, H.; Yamamoto, T. Effects of chronic administration of interferon alpha A/D on serotonergic receptors in rat brain. Neurochem Res. 1999, 24, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Kim, Y.K. Cytokine-serotonin interaction through IDO: A neurodegeneration hypothesis of depression. Med. Hypotheses 2003, 61, 519–525. [Google Scholar] [CrossRef]
- Berk, M.; Dean, O.; Drexhage, H.; McNeil, J.J.; Moylan, S.; O’Neil, A.; Davey, C.G.; Sanna, L.; Maes, M. Aspirin: A review of its neurobiological properties and therapeutic potential for mental illness. BMC Med. 2013, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Eyre, H.A.; Air, T.; Proctor, S.; Rositano, S.; Baune, B.T. A critical review of the efficacy of non-steroidal anti-inflammatory drugs in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 57, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.I.; Bosetti, F. Do lithium and anticonvulsants target the brain arachidonic acid cascade in bipolar disorder? Arch. Gen. Psychiatry 2002, 59, 592–596. [Google Scholar] [CrossRef]
- Müller, N. COX-2 Inhibitors, Aspirin, and Other Potential Anti-Inflammatory Treatments for Psychiatric Disorders. Front. Psychiatry 2019, 10, 375. [Google Scholar] [CrossRef] [Green Version]
- Sethi, R.; Gómez-Coronado, N.; Walker, A.J.; Robertson, O.D.; Agustini, B.; Berk, M.; Dodd, S. Neurobiology and Therapeutic Potential of Cyclooxygenase-2 (COX-2) Inhibitors for Inflammation in Neuropsychiatric Disorders. Front. Psychiatry 2019, 10, 605. [Google Scholar] [CrossRef]
- Famitafreshi, H.; Karimian, M. Prostaglandins as the Agents That Modulate the Course of Brain Disorders. Degener. Neurol. Neuromuscul. Dis. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Horrobin, D.F.; Ally, A.I.; Karmali, R.A.; Karmazyn, M.; Manku, M.S.; Morgan, R.O. Prostaglandins and schizophrenia: Further discussion of the evidence. Psychol. Med. 1978, 8, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Lieb, J.; Karmali, R.; Horrobin, D. Elevated levels of prostaglandin E2 and thromboxane B2 in depression. Prostaglandins Leukot. Med. 1983, 10, 361–367. [Google Scholar] [CrossRef]
- Qian, Z.; Ding, W.; Zhou, Q.; Sun, C.; Xu, K. Depression Induced by CUMS Leads to Bladder Cancer Development and Local Tumor Immunosuppression in Mice. J. Oncol. 2021, 2021, 5537523. [Google Scholar] [CrossRef] [PubMed]
- Onaka, Y.; Shintani, N.; Nakazawa, T.; Haba, R.; Ago, Y.; Wang, H.; Kanoh, T.; Hayata-Takano, A.; Hirai, H.; Nagata, K.Y.; et al. CRTH2, a prostaglandin D2 receptor, mediates depression-related behavior in mice. Behav. Brain. Res. 2015, 284, 131–137. [Google Scholar] [CrossRef]
- Guo, J.Y.; Li, C.Y.; Ruan, Y.P.; Sun, M.; Qi, X.L.; Zhao, B.S.; Luo, F. Chronic treatment with celecoxib reverses chronic unpredictable stress-induced depressive-like behavior via reducing cyclooxygenase-2 expression in rat brain. Eur. J. Pharmacol. 2009, 612, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, X.; Manku, M. Increased Phospholipase A2 Activity and Inflammatory Response But Decreased Nerve Growth Factor Expression in the Olfactory Bulbectomized Rat Model of Depression: Effects of Chronic Ethyl-Eicosapentaenoate Treatment. J. Neurosci. 2009, 29, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Feng, Y.B.; Wang, L.; Shen, J.; Li, Y.; Fan, C.; Wang, P.; Yu, S.Y. COX-2 inhibition rescues depression-like behaviors via suppressing glial activation, oxidative stress and neuronal apoptosis in rats. Neuropharmacology 2019, 160, 107779. [Google Scholar] [CrossRef]
- Kumar, A.; Kumari, B.; Kumar, P. Protective effects of selective and non-selective cyclooxygenase inhibitors in an animal model of chronic stress. Neurosci. Bull. 2010, 26, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Myint, A.M.; Steinbusch, H.W.; Goeghegan, L.; Luchtman, D.; Kim, Y.K.; Leonard, B.E. Effect of the COX-2 inhibitor celecoxib on behavioural and immune changes in an olfactory bulbectomised rat model of depression. Neuroimmunomodulation 2007, 14, 65–71. [Google Scholar] [CrossRef]
- Müller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Müller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: Results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry. 2006, 1, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.G.; Monkul, E.S.; Hatch, J.P.; Fonseca, M.; Zunta-Soares, G.B.; Frey, B.N.; Bowden, C.L.; Soares, J.C. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: A double-blind, randomized, placebo-controlled study. Hum. Psychopharmacol. 2008, 23, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry. 2014, 71, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Na, K.S.; Lee, K.J.; Lee, J.S.; Cho, Y.S.; Jung, H.Y. Efficacy of adjunctive celecoxib treatment for patients with major depressive disorder: A meta-analysis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 48, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Casolini, P.; Catalani, A.; Zuena, A.R.; Angelucci, L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J. Neurosci. Res. 2002, 68, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Wang, X.; Pace, T.W.; Wu, H.; Miller, A.H. Inhibition of COX-2 by celecoxib enhances glucocorticoid receptor function. Mol. Psychiatry 2005, 10, 426–428. [Google Scholar] [CrossRef] [Green Version]
- Fields, C.; Drye, L.; Vaidya, V.; Lyketsos, C.; ADAPT Research Group. Celecoxib or naproxen treatment does not benefit depressive symptoms in persons age 70 and older: Findings from a randomized controlled trial. Am. J. Geriatr. Psychiatry 2012, 20, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Uher, R.; Carver, S.; Power, R.A.; Mors, O.; Maier, W.; Rietschel, M.; Hauser, J.; Dernovsek, M.Z.; Henigsberg, N.; Souery, D.; et al. Non-steroidal anti-inflammatory drugs and efficacy of antidepressants in major depressive disorder. Psychol. Med. 2012, 42, 2027–2035. [Google Scholar] [CrossRef]
- Aid, S.; Langenbach, R.; Bosetti, F. Neuroinflammatory response to lipopolysaccharide is exacerbated in mice genetically deficient in cyclooxygenase-2. J. Neuroinflamm. 2008, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Aid, S.; Bosetti, F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie 2011, 93, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Fišar, Z.; Medina, M.; Scapagnini, G.; Nowak, G.; Berk, M. New drug targets in depression: Inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates—Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology 2012, 20, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Dudek, K.A.; Dion-Albert, L.; Kaufmann, F.N.; Tuck, E.; Lebel, M.; Menard, C. Neurobiology of resilience in depression: Immune and vascular insights from human and animal studies. Eur. J. Neurosci. 2021, 53, 183–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhallen, A.M.; Alonso-Martínez, S.; Renken, R.J.; Marsman, J.-B.C.; Ter Horst, G.J. Depressive symptom trajectory following romantic relationship breakup and effects of rumination, neuroticism and cognitive control. Stress Health, 2021; Epub Ahead of Print. [Google Scholar]
- Isella, C.; Gasparini, A.; Lucca, G.; Ielmini, M.; Caselli, I.; Poloni, N.; Dajelli Ermolli, C.; Caravati, F.; Castiglioni, B.; De Ponti, R.; et al. Resilience, Cardiological Outcome, and Their Correlations With Anxious-Depressive Symptoms and Quality of Life in Patients With an Implantable Cardioverter Defibrillator. Front. Psychiatry 2021, 12, 763726. [Google Scholar] [CrossRef]
- Russo, S.J.; Murrough, J.W.; Han, M.-H.; Charney, D.S.; Nestler, E.J. Neurobiology of resilience. Nat. Neurosci. 2012, 15, 1475–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzer, R.; Cohen, S.; Russo, S.J.; Dinan, T.G. Resilience and immunity. Brain Behav. Immun. 2018, 74, 28–42. [Google Scholar] [CrossRef]
- Levone, B.R.; Cryan, J.F.; O’Leary, O.F. Role of adult hippocampal neurogenesis in stress resilience. Neurobiol. Stress 2014, 1, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Huang, H.; Li, S.; Zhou, M.; Liu, Z.; Huang, R.; Liao, W.; Xie, P.; Zhou, J. Hippocampal proteomic changes of susceptibility and resilience to depression or anxiety in a rat model of chronic mild stress. Transl. Psychiatry 2019, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, A.; Segal, M.; Stork, O. Allostatic gene regulation of inhibitory synaptic factors in the rat ventral hippocampus in a juvenile/adult stress model of psychopathology. Eur. J. Neurosci. 2020; Epub Ahead of Print. [Google Scholar]
- Zhang, X.; Liu, Y.; Hong, X.; Li, X.; Meshul, C.K.; Moore, C.; Yang, Y.; Han, Y.; Li, W.-G.; Qi, X.; et al. NG2 glia-derived GABA release tunes inhibitory synapses and contributes to stress-induced anxiety. Nat. Commun. 2021, 12, 5740. [Google Scholar] [CrossRef]
- Willner, P.; Towell, A.; Sampson, D.; Sophokleous, S.; Muscat, R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology 1987, 93, 358–364. [Google Scholar] [CrossRef]
- Willner, P.; Belzung, C. Treatment-resistant depression: Are animal models of depression fit for purpose? Psychopharmacology 2015, 232, 3473–3495. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M. Development of a rating scale for primary depressive illness. Br. J. Soc. Clin. Psychol. 1967, 6, 278–296. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.F. Letter: Pathophysiology of depressive syndromes. Biol. Psychiatry 1974, 1, 119–120. [Google Scholar]
- Strekalova, T.; Spanagel, R.; Bartsch, D.; Henn, F.; Gass, P. Stress-induced anhedonia in mice is associated with deficits in forced swimming and exploration. Neuropsychopharmacology 2004, 29, 2007–2017. [Google Scholar] [CrossRef]
- Strekalova, T.; Steinbusch, H. Measuring behavior with chronic stress depression model in mice. Prog. Neuro-Psychopharm. Biol. Psychiatry 2010, 34, 348–361. [Google Scholar] [CrossRef]
- Cline, B.; Costa-Nunes, J.; Cespuglio, R.; Markova, N.; Santos, A.; Bukhman, Y.; Kubatiev, A.; Steinbusch, H.; Lesch, K.; Strekalova, T. Dicholine succinate, the neuronal insulin sensitizer, normalizes behavior, REM sleep, hippocampal pGSK3 beta and mRNAs of NMDA receptor subunits in mouse models of depression. Front. Behav. Neurosci. 2015, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Gorinski, N.; Bijata, M.; Prasad, S.; Wirth, A.; Abdel Galil, D.; Zeug, A.; Bazovkina, D.; Kondaurova, E.; Kulikova, E.; Ilchibaeva, T.; et al. Attenuated palmitoylation of serotonin receptor 5-HT1A affects receptor function and contributes to depression-like behaviors. Nat. Commun. 2019, 10, 3924. [Google Scholar] [CrossRef] [Green Version]
- Strekalova, T.; Couch, Y.; Kholod, N.; Boyks, M.; Malin, D.; Leprince, P.; Steinbusch, H.M. Update in the methodology of the chronic stress paradigm: Internal control matters. Behav. Brain Funct. 2011, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Strekalova, T.; Gorenkova, N.; Schunk, E.; Dolgov, O.; Bartsch, D. Selective effects of citalopram in the mouse model of stress-induced anhedonia with control effects for chronic stress. Behav. Pharmacology 2006, 17, 271–287. [Google Scholar] [CrossRef]
- Cline, B.H.; Anthony, D.C.; Lysko, A.; Dolgov, O.; Anokhin, K.; Schroeter, C.; Malin, D.; Kubatiev, A.; Steinbusch, H.W.; Lesch, K.-P.; et al. Lasting downregulation of the lipid peroxidation enzymes in the prefrontal cortex of mice susceptible to stress-induced anhedonia. Behav. Brain Res. 2015, 276, 118–129. [Google Scholar] [CrossRef]
- Couch, Y.; Anthony, D.; Dolgov, O.; Revischin, A.; Festoff, B.; Santos, A.; Steinbusch, H.; Strekalova, T. Microglial activation, increased TNF and SERT expression in the prefrontal cortex define stress behaviour in mice susceptible to anhedonia. Brain Behav. Immun. 2013, 29, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Vignisse, J.; Sambon, M.; Gorlova, A.; Pavlov, D.; Caron, N.; Malgrange, B.; Shevtsova, E.; Svistunov, A.; Anthony, D.; Markova, N.; et al. Thiamine and benfotiamine prevent stress-induced suppression of hippocampal neurogenesis in mice exposed to predation without affecting brain thiamine diphosphate levels. Mol. Cell. Neurosci. 2017, 82, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Costa-Nunes, J.; Zubareva, O.; Araújo-Correia, M.; Valença, A.; Schroeter, C.A.; Pawluski, J.L.; Vignisse, J.; Steinbusch, H.; Hermes, D.; Phillipines, M.; et al. Altered emotionality, hippocampus-dependent performance and expression of NMDA receptor subunit mRNAs in chronically stressed mice. Stress 2014, 17, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Strekalova, T.; Bahzenova, N.; Trofimov, A.; Schmitt-Böhrer, A.G.; Markova, N.; Grigoriev, V.; Zamoysk, V.; Serkova, T.; Redkozubova, O.; Vinogradova, D.; et al. Pro-neurogenic, Memory-Enhancing and Anti-stress Effects of DF302, a Novel Fluorine Gamma-Carboline Derivative with Multi-target Mechanism of Action. Mol. Neurobiol. 2018, 55, 335–349. [Google Scholar] [CrossRef]
- Markova, N.; Bazhenova, N.; Anthony, D.C.; Vignisse, J.; Svistunov, A.; Lesch, K.P.; Bettendorff, L.; Strekalova, T. Thiamine and benfotiamine improve cognition and ameliorate GSK-3β-associated stress-induced behaviours in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 148–156. [Google Scholar] [CrossRef]
- Pavlov, D.; Markova, N.; Bettendorff, L.; Chekhonin, V.; Pomytkin, I.; Lioudyno, V.; Svistunov, A.; Ponomarev, E.; Lesch, K.P.; Strekalova, T. Elucidating the functions of brain GSK3α: Possible synergy with GSK3β upregulation and reversal by antidepressant treatment in a mouse model of depressive-like behaviour. Behav. Brain Res. 2017, 335, 122–127. [Google Scholar] [CrossRef]
- Sang, N.; Zhang, J.; Marcheselli, V.; Bazan, N.G.; Chen, C. Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J. Neurosci. 2005, 25, 9858–9870. [Google Scholar] [CrossRef]
- Chen, C.; Bazan, N.G. Acetaminophen modifies hippocampal synaptic plasticity via a presynaptic 5-HT2 receptor. Neuroreport 2003, 14, 743–747. [Google Scholar] [CrossRef]
- Ito, N.; Hirose, E.; Ishida, T.; Hori, A.; Nagai, T.; Kobayashi, Y.; Kiyohara, H.; Oikawa, T.; Hanawa, T.; Odaguchi, H. Kososan, a Kampo medicine, prevents a social avoidance behavior and attenuates neuroinflammation in socially defeated mice. J. Neuroinflamm. 2017, 14, 98. [Google Scholar] [CrossRef]
- Cathomas, F.; Murrough, J.W.; Nestler, E.J.; Han, M.H.; Russo, S.J. Neurobiology of Resilience: Interface Between Mind and Body. Biol. Psychiatry 2019, 86, 410–420. [Google Scholar] [CrossRef]
- Kamimura, Y.; Kuwagaki, E.; Hamano, S.; Kobayashi, M.; Yamada, Y.; Takahata, Y.; Yoshimoto, W.; Morimoto, H.; Yasukawa, T.; Uozumi, Y.; et al. Reproducible induction of depressive-like behavior in C57BL/6J mice exposed to chronic social defeat stress with a modified sensory contact protocol. Life Sci. 2021, 282, 119821. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.B.; Pinna, G.; Barros, H.M.T. The Role of HPA Axis and Allopregnanolone on the Neurobiology of Major Depressive Disorders and PTSD. Int. J. Mol. Sci. 2021, 22, 5495. [Google Scholar] [CrossRef] [PubMed]
- Colasanto, M.; Madigan, S.; Korczak, D.J. Depression and inflammation among children and adolescents: A meta-analysis. J. Affect. Disord. 2020, 277, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Mazza, M.G.; De Lorenzo, R.; Conte, C.; Poletti, S.; Vai, B.; Bollettini, I.; Melloni, E.M.T.; Furlan, R.; Ciceri, F.; Rovere-Querini, P.; et al. Anxiety and depression in COVID-19 survivors: Role of inflammatory and clinical predictors. Brain Behav. Immun. 2020, 89, 594–600. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Busquets-Cortés, C.; Capó, X.; Tejada, S.; Tur, J.A.; Pons, A.; Sureda, A. Cyclooxygenase-2 Inhibitors as a Therapeutic Target in Inflammatory Diseases. Curr. Med. Chem. 2019, 26, 3225–3241. [Google Scholar] [CrossRef]
- Myint, A.M.; Schwarz, M.J.; Steinbusch, H.W.M.; Leonard, B.E. Neuropsychiatric disorders related to interferon and interleukins treatment. Metab. Brain Dis. 2009, 24, 55–68. [Google Scholar] [CrossRef]
- Tanaka, K.; Furuyashiki, T.; Kitaoka, S.; Senzai, Y.; Imoto, Y.; Segi-Nishida, E.; Deguchi, Y.; Breyer, R.M.; Breyer, M.D.; Narumiya, S. Prostaglandin E2-mediated attenuation of mesocortical dopaminergic pathway is critical for susceptibility to repeated social defeat stress in mice. J. Neurosci. 2012, 32, 4319–4329. [Google Scholar] [CrossRef] [Green Version]
- Savitz, J.; Frank, M.B.; Victor, T.; Bebak, M.; Marino, J.H.; Bellgowan, P.S.; McKinney, B.A.; Bodurka, J.; Kent Teague, T.; Drevets, W.C. Inflammation and neurological disease-related genes are differentially expressed in depressed patients with mood disorders and correlate with morphometric and functional imaging abnormalities. Brain Behav. Immun. 2013, 31, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N.; Okada, S. Cyclooxygenase-1 and -2 in spinally projecting neurons are involved in CRF-induced sympathetic activation. Auton. Neurosci. 2009, 151, 82–89. [Google Scholar] [CrossRef]
- Dargahi, L.; Nasiraei-Moghadam, S.; Abdi, A.; Khalaj, L.; Moradi, F.; Ahmadiani, A. Cyclooxygenase (COX)-1 activity precedes the COX-2 induction in Aβ-induced neuroinflammation. J. Mol. Neurosci. 2011, 45, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, B.M. FDA approves first celecoxib generics. JAMA 2014, 311, 2470. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Pariante, C.M. Trial failures of anti-inflammatory drugs in depression. Lancet Psychiatry 2020, 7, 837. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Gałecki, P.; Talarowska, M.; Bobińska, K.; Szemraj, J. COX-2 gene expression is correlated with cognitive function in recurrent depressive disorder. Psychiatry Res. 2014, 215, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.I.; Chaudhry, I.B.; Khoso, A.B.; Husain, M.O.; Hodsoll, J.; Ansari, M.A.; Naqvi, H.A.; Minhas, F.A.; Carvalho, A.F.; Meyer, J.H.; et al. Minocycline and celecoxib as adjunctive treatments for bipolar depression: A multicentre, factorial design randomised controlled trial. Lancet Psychiatry 2020, 7, 515–527. [Google Scholar] [CrossRef]
- Kalkman, H.O.; Feuerbach, D. Antidepressant therapies inhibit inflammation and microglial M1-polarization. Pharmacol. Ther. 2016, 163, 82–93. [Google Scholar] [CrossRef]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF-α and CRP in Elderly Patients with Depression or Alzheimer’s disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Strekalova, T.; Evans, M.; Chernopiatko, A.; Couch, Y.; Costa-Nunes, J.; Cespuglio, R.; Chesson, L.; Vignisse, J.; Steinbusch, H.W.; Anthony, D.C.; et al. Deuterium content of water increases depression susceptibility: The potential role of a serotonin-related mechanism. Behav. Brain Res. 2015, 277, 237–244. [Google Scholar] [CrossRef]
- Cline, B.H.; Steinbusch, H.W.; Malin, D.; Revishchin, A.V.; Pavlova, G.V.; Cespuglio, R.; Strekalova, T. The neuronal insulin sensitizer dicholine succinate reduces stress-induced depressive traits and memory deficit: Possible role of insulin-like growth factor 2. BMC Neurosci. 2012, 13, 110. [Google Scholar] [CrossRef] [Green Version]
- Vignisse, J.; Steinbusch, H.W.M.; Grigoriev, V.; Bolkunov, A.; Proshin, A.; Bettendorff, L.; Bachurin, S.; Strekalova, T. Concomitant manipulation of murine NMDA- and AMPA-receptors to produce pro-cognitive drug effects in mice. Eur. Neuropsychopharmacol. 2014, 24, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Couch, Y.; Trofimov, A.; Markova, N.; Nikolenko, V.; Steinbusch, H.; Chekhonin, V.; Schroeter, C.; Lesch, K.; Anthony, D.; Strekalova, T. Low-dose lipopolysaccharide (LPS) inhibits aggressive and augments depressive behaviours in a chronic mild stress model in mice. J. Neuroinflamm. 2016, 1, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strekalova, T.V.; Cespuglio, R.; Koval’zon, V.M. Depressive-like state and sleep in laboratory mice. Zh. Vyssh. Nerv. Deiat. Im. I P Pavlova 2008, 58, 728–737. [Google Scholar] [PubMed]
- Strekalova, T.; Spanagel, R.; Dolgov, O.; Bartsch, D. Stress-induced hyperlocomotion as a confounding factor in anxiety and depression models in mice. Behav. Pharmacol. 2005, 16, 171–180. [Google Scholar] [CrossRef]
- Veniaminova, E.; Cespuglio, R.; Cheung, C.W.; Umriukhin, A.; Markova, N.; Shevtsova, E.; Lesch, K.-P.; Anthony, D.C.; Strekalova, T. Autism-Like Behaviours and Memory Deficits Result from a Western Diet in Mice. Neural Plast. 2017, 2017, 9498247. [Google Scholar] [CrossRef] [Green Version]
- Malatynska, E.; Steinbusch, H.W.M.; Redkozubova, O.; Bolkunov, A.; Kubatiev, A.; Yeritsyan, N.B.; Vignisse, J.; Bachurin, S.; Strekalova, T. Anhedonic-like traits and lack of affective deficits in 18-month-old C57BL/6 mice: Implications for modeling elderly depression. Exp. Gerontol. 2012, 47, 552–564. [Google Scholar] [CrossRef]
- Costa-Nunes, J.P.; Cline, B.H.; Araújo-Correia, M.; Valença, A.; Markova, N.; Dolgov, O.; Kubatiev, A.; Yeritsyan, N.; Steinbusch, H.W.M.; Strekalova, T. Animal Models of Depression and Drug Delivery with Food as an Effective Dosing Method: Evidences from Studies with Celecoxib and Dicholine Succinate. Biomed Res. Int. 2015, 2015, 596126. [Google Scholar] [CrossRef]
- de Munter, J.; Pavlov, D.; Gorlova, A.; Sicker, M.; Proshin, A.; Kalueff, A.V.; Svistunov, A.; Kiselev, D.; Nedorubov, A.; Morozov, S.; et al. Increased Oxidative Stress in the Prefrontal Cortex as a Shared Feature of Depressive- and PTSD-Like Syndromes: Effects of a Standardized Herbal Antioxidant. Front. Nutr. 2021, 8, 661455. [Google Scholar] [CrossRef]
- Costa-Nunes, J.P.; Gorlova, A.; Pavlov, D.; Cespuglio, R.; Gorovaya, A.; Proshin, A.; Umriukhin, A.; Ponomarev, E.D.; Kalueff, A.V.; Strekalova, T.; et al. Ultrasound stress compromises the correlates of emotional-like states and brain AMPAR expression in mice: Effects of antioxidant and anti-inflammatory herbal treatment. Stress 2020, 23, 481–495. [Google Scholar] [CrossRef]
- Veniaminova, E.; Oplatchikova, M.; Bettendorff, L.; Kotenkova, E.; Lysko, A.; Vasilevskaya, E.; Kalueff, A.V.; Fedulova, L.; Umriukhin, A.; Lesch, K.P.; et al. Prefrontal cortex inflammation and liver pathologies accompany cognitive and motor deficits following Western diet consumption in non-obese female mice. Life Sci. 2020, 241, 117163. [Google Scholar] [CrossRef]
- Pavlov, D.; Bettendorff, L.; Gorlova, A.; Olkhovik, A.; Kalueff, A.V.; Ponomarev, E.D.; Inozemtsev, A.; Chekhonin, V.; Lesch, K.P.; Anthony, D.C.; et al. Neuroinflammation and aberrant hippocampal plasticity in a mouse model of emotional stress evoked by exposure to ultrasound of alternating frequencies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 90, 104–116. [Google Scholar] [CrossRef] [PubMed]
- David, D.J.; Gourion, D. Antidépresseurs et tolérance: Déterminants et prise en charge des principaux effets indésirables [Antidepressant and tolerance: Determinants and management of major side effects]. Encephale 2016, 42, 553–561. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strekalova, T.; Pavlov, D.; Trofimov, A.; Anthony, D.C.; Svistunov, A.; Proshin, A.; Umriukhin, A.; Lyundup, A.; Lesch, K.-P.; Cespuglio, R. Hippocampal Over-Expression of Cyclooxygenase-2 (COX-2) Is Associated with Susceptibility to Stress-Induced Anhedonia in Mice. Int. J. Mol. Sci. 2022, 23, 2061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042061
Strekalova T, Pavlov D, Trofimov A, Anthony DC, Svistunov A, Proshin A, Umriukhin A, Lyundup A, Lesch K-P, Cespuglio R. Hippocampal Over-Expression of Cyclooxygenase-2 (COX-2) Is Associated with Susceptibility to Stress-Induced Anhedonia in Mice. International Journal of Molecular Sciences. 2022; 23(4):2061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042061
Chicago/Turabian StyleStrekalova, Tatyana, Dmitrii Pavlov, Alexander Trofimov, Daniel C. Anthony, Andrei Svistunov, Andrey Proshin, Aleksei Umriukhin, Alexei Lyundup, Klaus-Peter Lesch, and Raymond Cespuglio. 2022. "Hippocampal Over-Expression of Cyclooxygenase-2 (COX-2) Is Associated with Susceptibility to Stress-Induced Anhedonia in Mice" International Journal of Molecular Sciences 23, no. 4: 2061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042061