2.1. Peptide Design and Physico-Chemical Properties
The first step in the activity and selectivity of cationic AMPs is the electrostatic interaction with the negatively charged bacterial membrane [
23,
24,
25]. Thus, different strategies are used to improve the antibacterial activity with the aim to create more efficient potential agents in clinical. The most-used approach is to find the correct balance between the net positive charge and the amphipathic properties to obtain peptides that can fold in α-helices or β-structures [
22,
26,
27,
28]. In the present study, we designed small peptides derived from the sequence of KHS-Cnd [
22]. KHS-Cnd was chosen because of its high antimicrobial activity against Gram-positive, Gram-negative, and multidrug-resistant bacteria and relatively low cytotoxicity. We synthesized three small peptides of 11 amino acids from the parental peptide. The sequences and physicochemical properties of the three derivatives corresponding to the N-terminal, central, and C-terminal portions of KHS-Cnd, named Pep-A, Pep-B, and Pep-C, are reported in
Table 1. Many studies report that the functionalization with fatty acid chains increases antifungal activity [
20,
21]. Thus, we modified the N-terminal by introducing a hydrophobic myristoyl moiety (14-carbon fatty acid) to favor the vehiculation and interaction of peptides with membranes and enhance their membrane-anchoring properties [
29,
30]. This modification increased the hydrophobicity of peptides without varying the peptide sequence.
Physico-chemical properties of myristoylated peptides, called Myr-A, Myr-B, and Myr-C, are also reported in
Table 1.
2.3. Membrane Partition Studies
To quantify the interaction of different lipidated and non-lipidated peptides with membrane mimetic systems, we measured the intrinsic fluorescence of Trp-1 upon partition with LUVs (large unilamellar vesicles) of different compositions. Briefly, the mixtures of POPC/POPG (70%/30%
w/
w) LUVs were used to mimic anionic membranes and POPC only to mimic zwitterionic membranes [
34,
35,
36]. LUVs do not entirely describe the complexity of natural membranes; still, they are used here to evaluate the preference of different peptides for anionic (bacteria and fungi) and zwitterionic (mammalian cell) membranes. The mole fraction partition constants
Kx were calculated from the partition isotherms as previously described [
37].
Figure 2 reports the partition isotherms for the titration of myristoylated and non-myristoylated peptides with POPC and POPC/POPG vesicles.
The values of
Kx for all peptides are reported in
Table 2 together with Δ
G values, calculated using the expression Δ
G = −RT lnKx, and the selectivity ratio defined as the ratio between partition constants determined in anionic and zwitterionic membranes. Higher values of the selectivity ratio represent a preference of the peptide for anionic vesicles, while lower values indicate a preference for the zwitterionic one.
Partitioning data suggest that myristoylated peptides interact with LUVs with higher affinity. The mole fraction partition coefficients Kx range from 3.0 × 104 to 5.2 × 104 with calculated Gibbs free energies of the partition of ~(−26) kJ/mol for non-myristoylated peptides, suggesting poor binding to membranes. Myristoylated peptides showed larger Kx values ranging from 1.0 × 105 in POPC to 1.7 × 106 POPC/POPG vesicles, indicating a stronger interaction with LUVs.
Moreover, myristoylated peptides prefer to interact with anionic membranes as described by the values of the selectivity ratio ranging between ~2 and 4. This feature should increase the antifungal activity, as described later. Mc Laughlin and colleagues stated that the presence of the myristoyl moiety is required to interact with membranes but is not always sufficient [
38]. The higher propensity of myristoylated peptides to interact with membranes can be explained by the presence of the myristoyl group and charge effects. Mainly, the values of Gibbs free energy ~(−33) kJ/mol are similar to that measured for the insertion of myristate into the bilayer [
39]. Thus, the synergism between the myristate insertion into the bilayer and the electrostatic interaction between the lysine residues present in the peptides and the anionic lipids can be the driving force of the process [
40].
2.4. Fluorescence Quenching Experiments
Peptides’ topology in the absence and presence of lipid vesicles has been evaluated by monitoring the quenching of Trp-1 by acrylamide. Acrylamide is a small neutral molecule that quenches the tryptophan residue with high efficiency [
41]. Quenching experiments were carried out in buffer and lipid vesicles as previously described [
42]. Typical results are shown in
Figure 3 for myristoylated and non-myristoylated peptides.
In the buffer, all non-myristoylated peptides and myristoylated peptides Myr-A and Myr-C showed typical linear behavior of the fluorescence intensity ratio in the absence (
Fo) and in the presence (
F) of the quencher vs. the quencher concentration. The same behavior was detected for myristoylated peptides in POPC and POPC/POPG lipid vesicles. The classical Stern–Volmer equation [
43]
was used to describe the fluorescence quenching, where
KSV is the Stern–Volmer or collisional constant, while
Fo and
F are the fluorescence in the absence and presence of the acrylamide Q, respectively. Values of
KSV are indicative of the dynamic quenching and are higher in buffer solution than in the presence of lipid vesicles (
Table 3), suggesting that Trp-1 is less accessible to the quencher in the presence of lipid vesicles and confirming the partition into the lipid bilayer.
As reported in
Figure 3, Myr-B in the buffer and non-myristoylated peptides in the presence of lipid vesicles showed non-linear behavior, with downward curvature at high quencher concentrations. This effect can be ascribed to selective quenching [
44] due to the presence of two different fluorophore populations (
a and
b) with a diverse solvent exposition: one more accessible to the quencher and the other less. The fluorescence intensity can be written as:
The fraction of total fluorescence has been determined using the following equation [
43]:
where
fa is the fraction of the accessible fluorophore at an infinite concentration of the quencher and
Ka is an average Stern–Volmer constant for the accessible fluorophore both exposed and buried. A plot of,
yields a straight line from which we determined the fitting parameters
fa and
Ka (
Figures S3 and S4). Values of
fa for non-myristoylated peptides in the presence of lipid vesicles are between 0.40 and 0.75, indicating two fluorophore populations, one accessible to the quencher and the other deeply buried in the phospholipid bilayer.
Furthermore, we calculated the NAF, defined as the ratio between the Stern–Volmer constant in the presence and absence of LUV [
45]:
Low values of NAF suggest protection of the fluorophore from the quencher and strong interaction with lipids. NAF values reported in
Table 3 are lower for myristoylated peptides than for non-myristoylated, suggesting a better interaction with the lipid vesicles. Peptide Myr-B has a lower value of NAF (0.07), indicating a strong interaction with the lipid and a preference for anionic lipid vesicles. Non-myristoylated peptides show high values of NAF, suggesting a weak interaction with lipid vesicles. All the data agree with the results obtained from the partition studies.
2.6. Molecular Dynamics Simulations
To investigate more deeply, with an atomic resolution, the interaction between the peptides and the membrane models, we carried out a series of molecular dynamics simulations of peptides in different environments, including water, TFE/water (50% v/v), and in the presence of POPC and POPC/POPG lipid bilayers.
The initial structure of peptides used for the MD in the solution was obtained using the I-Tasser bioinformatics tool, which suggested, for all peptides, an α-helix conformation.
The MD simulations in water of all peptides showed a rapid loss of the initial α-helix conformation (
Figure S5). Only for Myr-B was the formation of a transient α-helix observed during 300 ns of simulation. Analysis of the peptide’s secondary structure as a function of the simulation time suggests that all peptides are unstructured with high conformational flexibility in the water, in agreement with the experimental results obtained by CD spectra.
In the presence of TFE/water (50%
v/
v), a less polar solvent with respect to water, Pep-B, Pep-C, and Myr-C conserved the α-helix conformation for 300 ns with a helicity of about 45%, 64%, and 82%, respectively (
Figure S5). In the case of Pep-A and Myr-A, the analysis of the secondary structure showed an unfolding of peptides after 120 ns and 220 ns of simulation (
Figure S5A,B).
We also simulated the adsorption of peptides on the surface of anionic and zwitterionic membrane models. The structures obtained at the end of the MD simulations in TFE/water were used as the initial conformation of peptides for the simulations in the presence of POPC and POPC/POPG lipid bilayers. The peptides were positioned close to the surface of the membranes with the principal axis parallel to the lipid surface and a distance of 1.0 nm between the center of mass of the peptide and the phosphorous atoms of lipids.
Figure 5 shows the representative snapshots of MD simulation at 1 μs of myristoylated peptides in the presence of POPC and POPC/POPG lipid bilayers. Myr-A and Myr-B interact with the surface of the zwitterionic and anionic membrane through an unfolded conformation, while Myr-C shows a higher tendency with respect to the other myristoylated peptide to fold in the presence of lipid bilayers. In particular, analysis of the time evolution of the secondary structure of Myr-C in POPC/POPG shows that the peptide conserves helicity (α-helix and π-helix) of about 45% (five residues) during the simulation duration (
Figure S6). On the other hand, in the presence of POPC, Myr-C interacts with the membrane, adopting, in the last 500 ns of simulation, a 3
10-helix with helicity of about 45.5% (
Figure S6).
The adsorption of myristoylated peptides to the surface of the lipid bilayer is driven mainly by the alkyl chain, linked to the N-terminal residue of peptides, and by the presence of positively charged amino acids. The alkyl chain penetrates in the hydrophobic region of the lipid bilayer while the positively charged amino acids stabilize the peptide–membrane interaction by forming hydrogen bonds and electrostatic interactions with lipid molecules. The peptide Myr-A in POPC shows a deep penetration of the myristoyl chain and Trp-1 in the hydrophobic region of the membrane (
Figure 5A and
Figure S7A). In particular, the average position of Trp-1 is located slightly below the position of the carbonyl group (CO
sn-1 and CO
sn-2) of POPC, as evidenced by the analysis of the average density distribution calculated to the lipid bilayer center (
Figure S7A). In POPC/POPG (
Figure 5B and
Figure S7B), the residues of Trp-1 and Lys-4 are located close to the lipid/water surface. The residue Arg-7 interacts with phosphate groups forming an arginine-phosphate salt bridge. The different penetrations into the lipid bilayer of POPC and POPC/POPG observed by MD simulation for Trp-1 of the peptide Myr-A is in agreement with the NAF values obtained by fluorescence-quenching experiments. In POPC/POPG, Trp-1 is close to the lipid surface and then more exposed to the quencher molecules with respect to POPC, where the aromatic residue can penetrate the hydrophobic region of the membrane.
The peptides Myr-B and Myr-C adsorbed on the surface of the lipid membrane show a different orientation with respect to Myr-A. The peptide Myr-B, in the presence of POPC and POPC/POPG, adopts an extended conformation parallel to the lipid surface with charged residues that interact with phosphate groups of lipids, and the residue Trp-1 located in the hydrophobic region of the membrane (
Figure 5C,D). Myr-C, which adopts a folding conformation (5–6 residues) when adsorbed by the membrane, in the presence of POPC shows residues of Trp-1 and all lysine residues except Lys-11 located inside the membrane close to the carbonyl group of lipids (
Figure 5E and
Figure S7E). On the contrary, in POPC/POPG, all charged amino acids of the peptide Myr-C are located close to the phosphate group (
Figure 5F and
Figure S5F) and interact with lipid molecules via the salt bridge and hydrogen bonds. In the case of Myr-B and Myr-C, analysis of the density distribution calculated with respect to the lipid bilayer center suggests that in POPC/POPG, the residue of Trp-1 is located more inside the membrane with respect to POPC, in agreement with the factor NAF obtained by fluorescence experiments.
The analysis of MD trajectories of non-myristoylated peptides in POPC and POPC/POPG reveals a lower affinity of peptides for lipid bilayers with respect to myristoylated peptides, as shown by the partition constants
Kx reported in
Table 2. In particular, the peptides Pep-B and Pep-C are adsorbed on the lipid surface and cannot deeply penetrate the membrane but lie on the surface, with the non-charged amino acids oriented toward the water phase (
Figures S8 and S9).
Figure 6 shows, for example, the different penetrations of Pep-C and Myr-C adsorbed on the lipid surface of POPC/POPG.
The density distribution of Trp-1 atoms, calculated with respect to the lipid bilayer center (
Figure S8), shows that for all non-myristoylated peptides, Trp-1 does not penetrate the hydrophobic portion of the membrane but remains located close to phosphate groups of lipids at the lipid/water interface. At the lipid/water interface, the residue Trp-1 is more exposed to water molecules and all molecular species present in the water phase. MD simulation results suggest that Trp-1 of myristoylated peptides is deeply immersed in the hydrophobic region of the membrane and, therefore, poorly exposed to water, with respect to non-myristoylated peptides. These results are in good agreement with the fluorescence experiments.
The MD simulations evidenced that the presence of the myristoyl moiety linked to Pep-A, Pep-B, and Pep-C deeply influences the properties of peptides, particularly their structure and affinity for lipid bilayers.
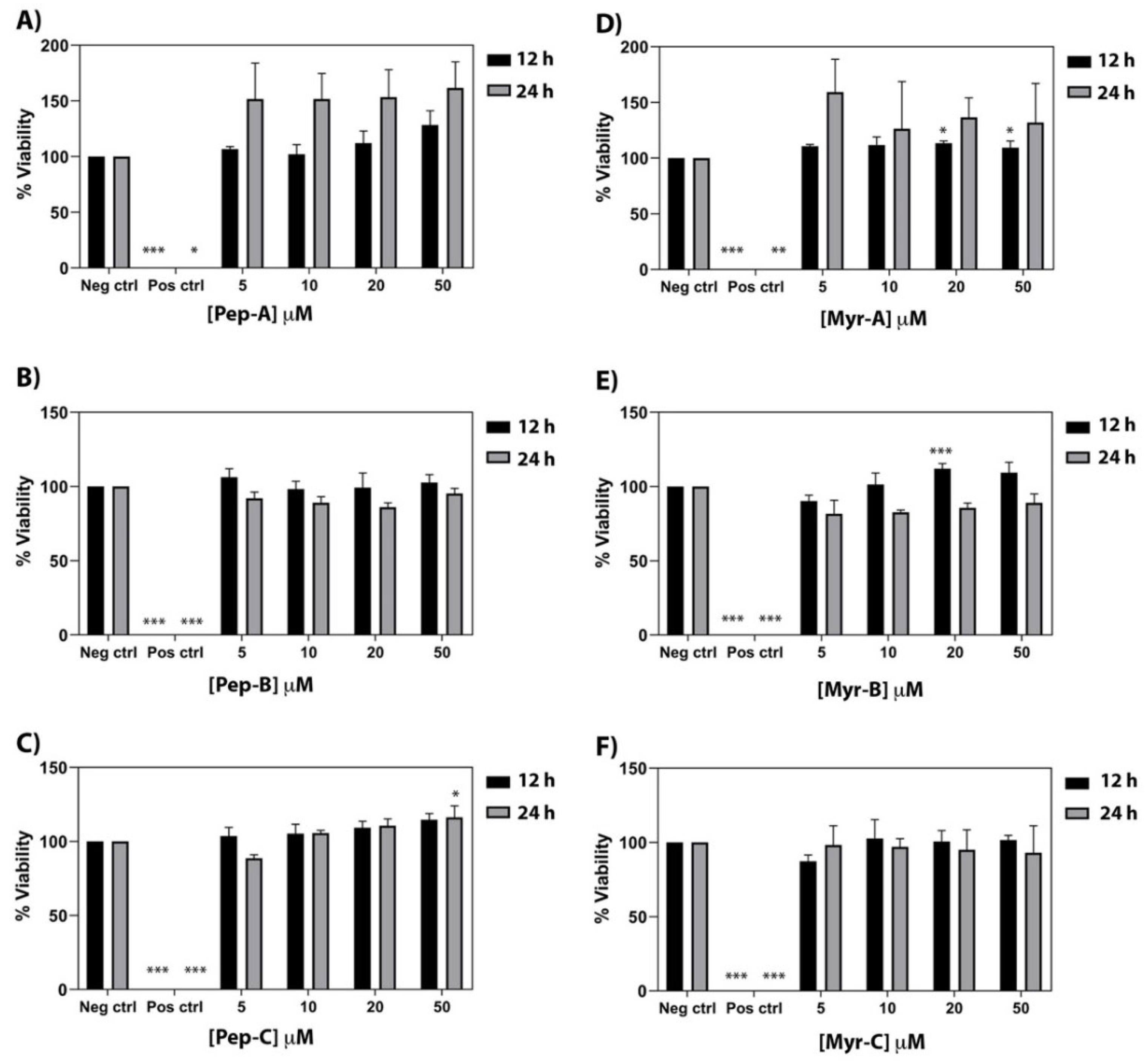
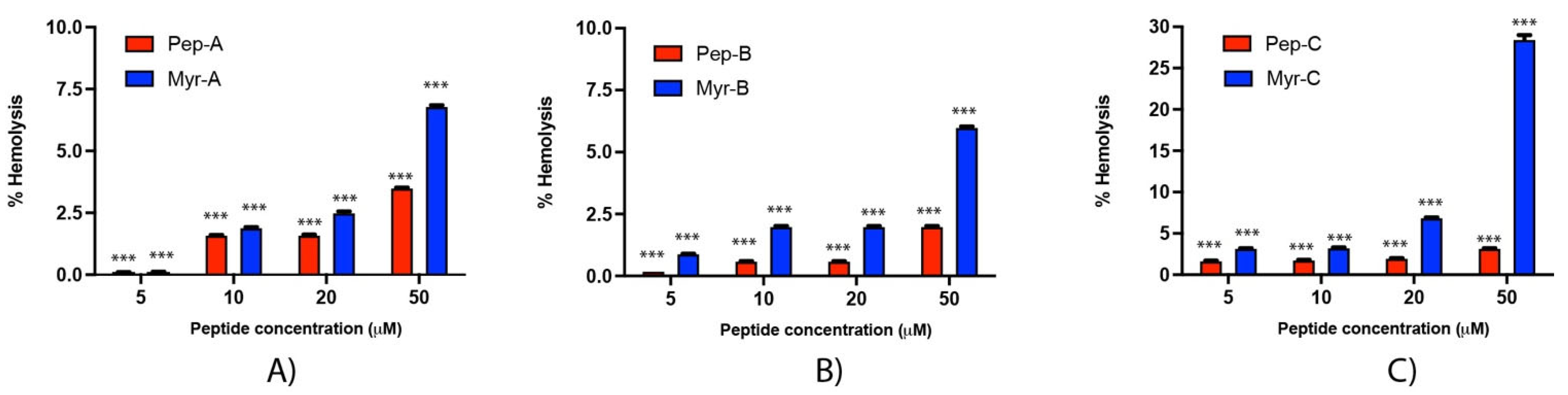
2.7. Antifungal Activity
The MIC
90 values of non-myristoylated and myristoylated peptides against different
Candida spp. are summarized in
Table 4. All lipopeptides have much lower MIC
90 values than their unfunctionalized counterparts, with the sole exception of the Myr-A peptide that maintains an unchanged MIC
90 value towards
C. auris. As already observed in the literature, these results confirm that increasing hydrophobicity by fatty acylation of AMPs with myristic acid enhances their antimicrobial activity and, in particular, the antifungal properties [
48]. The evidenced MIC
90 values are between 5 and 21 µM for all selected
Candida species, while only
Candida auris showed a higher range with Myr-B, which is the most active (MIC
90 20 µM). Myr-A and Myr-C were the most active against the selected
Candida species, and Myr-B particularly showed a MIC of 8 μg/mL (10 μM) vs.
C. tropicalis. Values of the single MICs of the 10 isolates for
Candida species are reported in the
Supplementary Information (Table S1). MIC values are higher than CMC, so MIC values are determined in the presence of lipopetide micellar aggregates and not the free monomer. Under these conditions, lipopetides are still soluble even if in the form of micellar aggregates. Thus, the activity of lipopetides might also depend on the so-called micellar mechanism, consisting of the solubilization of part of the host membrane into mixed micelles. However, further studies are needed to verify this hypothesis.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}