
Evaluation of Myeloperoxidase as Target for Host-Directed Therapy in Tuberculosis In Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Mycobacterial Burden and Disease Manifestations in M. tuberculosis Infected C3Heb/FeJ Mice Remain Unaffected by ABAH Treatment
2.2. Quantitative Characterization of Cellular Compositions and Effector Functions in Lungs of M. tuberculosis Infected Mice with or without ABAH Treatment
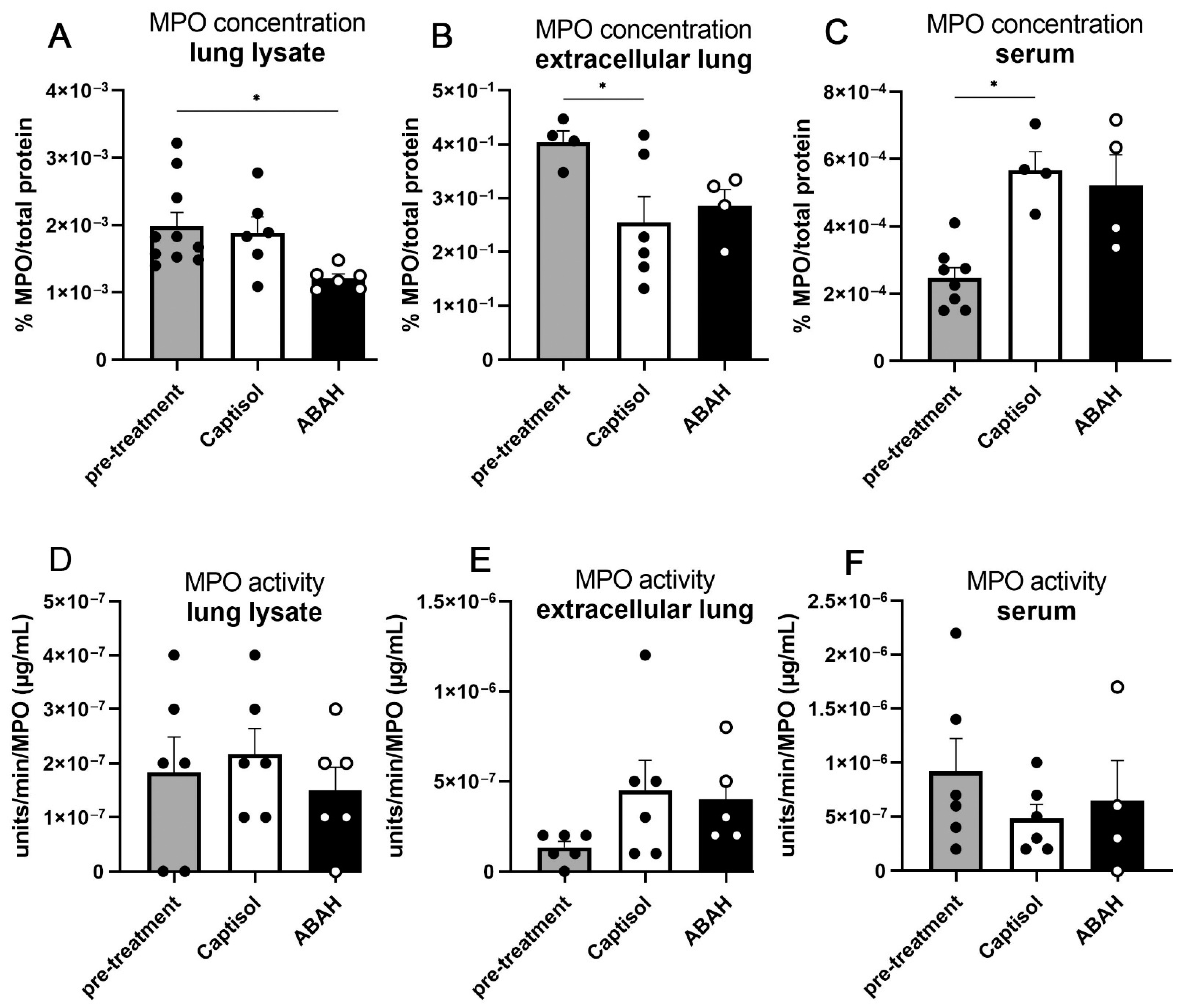
2.3. Necrosis and MPO within M. tuberculosis Lungs Were Not Affected by ABAH Treatment
2.4. Slight Reductions in Proinflammatory Cytokine Responses in ABAH-Treated Lungs
2.5. Co-Treatment with ABAH and Isoniazid
2.6. MPO Inhibition Did Not Reduce ROS Production and Necrosis in Infected Murine Neutrophils In Vitro
3. Discussion
4. Materials and Methods
4.1. Ethical Statements
4.2. Isolation of Human Neutrophils
4.3. M. tuberculosis Culture
4.4. Infections and Treatment
4.5. Necrosis Assays
4.6. Colony-Forming Unit Assay
4.7. Histology
4.8. Flow Cytometry
4.9. MPO Concentration and Activity Assays
4.10. Cytokine Measurement
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABAH | 4-Aminobenzoic acid hydrazide |
| DHR123 | dihydrorhodamine 123 |
| INH | isoniazid |
| iNOS | inducible NO synthase |
| LDH | lactate dehydrogenase |
| MDR/XDR | multi and extensively drug-resistant |
| MPO | myeloperoxidase |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
References
- Adams, L.B.; Dinauer, M.C.; Morgenstern, D.E.; Krahenbuhl, J.L. Comparison of the roles of reactive oxygen and nitrogen intermediates in the host response to Mycobacterium tuberculosis using transgenic mice. Tuber. Lung Dis. 1997, 78, 237–246. [Google Scholar] [CrossRef]
- Baghaei, P.; Tabarsi, P.; Dorriz, D.; Marjani, M.; Shamaei, M.; Pooramiri, M.V.; Mansouri, D.; Farnia, P.; Masjedi, M.; Velayati, A. Adverse effects of multidrug-resistant tuberculosis treatment with a standardized regimen: A report from Iran. Am. J. Ther. 2011, 18, e29–e34. [Google Scholar] [CrossRef]
- Balaiya, S.; Chalam, K.V. An In vitro Assay to Quantify Nitrosative Component of Oxidative Stress. J. Mol. Genet. Med. 2014, 8, 120. [Google Scholar]
- Berry, M.P.; Graham, C.M.; Mcnab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohrer, A.C.; Castro, E.; Hu, Z.; Queiroz, A.T.L.; Tocheny, C.E.; Assmann, M.; Sakai, S.; Nelson, C.; Baker, P.J.; Ma, H.; et al. Eosinophils are part of the granulocyte response in tuberculosis and promote host resistance in mice. J. Exp. Med. 2021, 218, e20210469. [Google Scholar] [CrossRef]
- Chen, S.; Chen, H.; Du, Q.; Shen, J. Targeting Myeloperoxidase (MPO) Mediated Oxidative Stress and Inflammation for Reducing Brain Ischemia Injury: Potential Application of Natural Compounds. Front. Physiol. 2020, 11, 433. [Google Scholar] [CrossRef] [PubMed]
- Corleis, B.; Korbel, D.; Wilson, R.; Bylund, J.; Chee, R.; Schaible, U.E. Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell Microbiol. 2012, 14, 1109–1121. [Google Scholar] [CrossRef]
- Crow, J.P. Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: Implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide 1997, 1, 145–157. [Google Scholar] [CrossRef]
- D’elia, R.V.; Harrison, K.; Oyston, P.C.; Lukaszewski, R.A.; Clark, G.C. Targeting the “Cytokine Storm” for Therapeutic Benefit. Clin. Vaccine Immunol. 2013, 20, 319–327. [Google Scholar] [CrossRef]
- Dallenga, T.; Linnemann, L.; Paudyal, B.; Repnik, U.; Griffiths, G.; Schaible, U.E. Targeting neutrophils for host-directed therapy to treat tuberculosis. Int. J. Med. Microbiol. 2018, 308, 142–147. [Google Scholar] [CrossRef]
- Dallenga, T.; Repnik, U.; Corleis, B.; Eich, J.; Reimer, R.; Griffiths, G.W.; Schaible, U.E.M. tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe 2017, 22, 519–530.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallenga, T.; Schaible, U.E. Neutrophils in tuberculosis--first line of defence or booster of disease and targets for host-directed therapy? Pathog. Dis. 2016, 74, ftw012. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; Griffin, J.D. Granulocyte colony-stimulating factor and its receptor. Blood 1991, 78, 2791–2808. [Google Scholar] [CrossRef] [Green Version]
- Domaszewska, T.; Scheuermann, L.; Hahnke, K.; Mollenkopf, H.; Dorhoi, A.; Kaufmann, S.H.E.; Weiner, J. Concordant and discordant gene expression patterns in mouse strains identify best-fit animal model for human tuberculosis. Sci. Rep. 2017, 7, 12094. [Google Scholar] [CrossRef] [PubMed]
- Driver, E.R.; Ryan, G.J.; Hoff, D.R.; Irwin, S.M.; Basaraba, R.J.; Kramnik, I.; Lenaerts, A.J. Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3181–3195. [Google Scholar] [CrossRef] [Green Version]
- Etna, M.P.; Giacomini, E.; Severa, M.; Coccia, E.M. Pro- and anti-inflammatory cytokines in tuberculosis: A two-edged sword in TB pathogenesis. Semin. Immunol. 2014, 26, 543–551. [Google Scholar] [CrossRef]
- Eum, S.Y.; Kong, J.H.; Hong, M.S.; Lee, Y.J.; Kim, J.H.; Hwang, S.H.; Cho, S.N.; Via, L.E.; Barry, C.E., 3rd. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 2010, 137, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Forghani, R.; Kim, H.J.; Wojtkiewicz, G.R.; Bure, L.; Wu, Y.; Hayase, M.; Wei, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase propagates damage and is a potential therapeutic target for subacute stroke. J. Cereb. Blood Flow Metab. 2015, 35, 485–493. [Google Scholar] [CrossRef]
- Forghani, R.; Wojtkiewicz, G.R.; Zhang, Y.; Seeburg, D.; Bautz, B.R.; Pulli, B.; Milewski, A.R.; Atkinson, W.L.; Iwamoto, Y.; Zhang, E.R.; et al. Demyelinating diseases: Myeloperoxidase as an imaging biomarker and therapeutic target. Radiology 2012, 263, 451–460. [Google Scholar] [CrossRef]
- Hoffmann, H.; Kohl, T.A.; Hofmann-Thiel, S.; Merker, M.; Beckert, P.; Jaton, K.; Nedialkova, L.; Sahalchyk, E.; Rothe, T.; Keller, P.M.; et al. Delamanid and Bedaquiline Resistance in Mycobacterium tuberculosis Ancestral Beijing Genotype Causing Extensively Drug-Resistant Tuberculosis in a Tibetan Refugee. Am. J. Respir. Crit. Care Med. 2016, 193, 337–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Wei, Y.; Lee, J.Y.; Wu, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase Inhibition Increases Neurogenesis after Ischemic Stroke. J. Pharmacol. Exp. Ther. 2016, 359, 262–272. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Wei, Y.; Wojtkiewicz, G.R.; Lee, J.Y.; Moskowitz, M.A.; Chen, J.W. Reducing myeloperoxidase activity decreases inflammation and increases cellular protection in ischemic stroke. J. Cereb. Blood Flow Metab. 2019, 39, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Kimmey, J.M.; Huynh, J.P.; Weiss, L.A.; Park, S.; Kambal, A.; Debnath, J.; Virgin, H.W.; Stallings, C.L. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015, 528, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Kramnik, I.; Beamer, G. Mouse models of human TB pathology: Roles in the analysis of necrosis and the development of host-directed therapies. Semin. Immunopathol. 2016, 38, 221–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macmicking, J.D.; North, R.J.; Lacourse, R.; Mudgett, J.S.; Shah, S.K.; Nathan, C.F. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 1997, 94, 5243–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckenzie, S.J.; Baker, M.S.; Buffinton, G.D.; Doe, W.F. Evidence of oxidant-induced injury to epithelial cells during inflammatory bowel disease. J. Clin. Investig. 1996, 98, 136–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, D. Control of granulocytes and macrophages: Molecular, cellular, and clinical aspects. Science 1991, 254, 529–533. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Mutlu, G.M.; Budinger, G.R.; Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013, 49, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, B.B.; Lovewell, R.R.; Olive, A.J.; Zhang, G.; Wang, W.; Eugenin, E.; Smith, C.M.; Phuah, J.Y.; Long, J.E.; Dubuke, M.L.; et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat. Microbiol. 2017, 2, 17072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat. Commun. 2020, 11, 5566. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Teixeira, L.; Tabone, O.; Graham, C.M.; Singhania, A.; Stavropoulos, E.; Redford, P.S.; Chakravarty, P.; Priestnall, S.L.; Suarez-Bonnet, A.; Herbert, E.; et al. Mouse transcriptome reveals potential signatures of protection and pathogenesis in human tuberculosis. Nat. Immunol. 2020, 21, 464–476. [Google Scholar] [CrossRef]
- Ohnishi, S.; Murata, M.; Kawanishi, S. DNA damage induced by hypochlorite and hypobromite with reference to inflammation-associated carcinogenesis. Cancer Lett. 2002, 178, 37–42. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Parida, S.K.; Axelsson-Robertson, R.; Rao, M.V.; Singh, N.; Master, I.; Lutckii, A.; Keshavjee, S.; Andersson, J.; Zumla, A.; Maeurer, M. Totally drug-resistant tuberculosis and adjunct therapies. J. Intern. Med. 2015, 277, 388–405. [Google Scholar] [CrossRef]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Pulli, B.; Ali, M.; Forghani, R.; Schob, S.; Hsieh, K.L.; Wojtkiewicz, G.; Linnoila, J.J.; Chen, J.W. Measuring myeloperoxidase activity in biological samples. PLoS ONE 2013, 8, e67976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rausch, P.G.; Moore, T.G. Granule enzymes of polymorphonuclear neutrophils: A phylogenetic comparison. Blood 1975, 46, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Saini, R.; Singh, S. Inducible nitric oxide synthase: An asset to neutrophils. J. Leukoc. Biol. 2019, 105, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Simms, P.E.; Ellis, T.M. Utility of flow cytometric detection of CD69 expression as a rapid method for determining poly- and oligoclonal lymphocyte activation. Clin. Diagn. Lab. Immunol. 1996, 3, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef]
- Ten, R.M.; Pease, L.R.; Mckean, D.J.; Bell, M.P.; Gleich, G.J. Molecular cloning of the human eosinophil peroxidase. Evidence for the existence of a peroxidase multigene family. J. Exp. Med. 1989, 169, 1757–1769. [Google Scholar] [CrossRef]
- Van Zandbergen, G.; Hermann, N.; Laufs, H.; Solbach, W.; Laskay, T. Leishmania promastigotes release a granulocyte chemotactic factor and induce interleukin-8 release but inhibit gamma interferon-inducible protein 10 production by neutrophil granulocytes. Infect. Immun. 2002, 70, 4177–4184. [Google Scholar] [CrossRef] [Green Version]
- Vilaplana, C.; Marzo, E.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.J. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J. Infect. Dis. 2013, 208, 199–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Taneja, R.; Razavi, H.M.; Law, C.; Gillis, C.; Mehta, S. Specific role of neutrophil inducible nitric oxide synthase in murine sepsis-induced lung injury in vivo. Shock 2012, 37, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Smith, S.D.; García-Cardeña, G.; Nathan, C.F.; Weiss, R.M.; Sessa, W.C. Bacterial infection induces nitric oxide synthase in human neutrophils. J. Clin. Investig. 1997, 99, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Whiteman, M.; Jenner, A.; Halliwell, B. Hypochlorous acid-induced base modifications in isolated calf thymus DNA. Chem. Res. Toxicol. 1997, 10, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Spencer, J.P.; Jenner, A.; Halliwell, B. Hypochlorous acid-induced DNA base modification: Potentiation by nitrite: Biomarkers of DNA damage by reactive oxygen species. Biochem. Biophys. Res. Commun. 1999, 257, 572–576. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2021; WHO Press: Genova, Italy, 2021. [Google Scholar]
- Yang, T.W.; Park, H.O.; Jang, H.N.; Yang, J.H.; Kim, S.H.; Moon, S.H.; Byun, J.H.; Lee, C.E.; Kim, J.W.; Kang, D.H. Side effects associated with the treatment of multidrug-resistant tuberculosis at a tuberculosis referral hospital in South Korea: A retrospective study. Medicine 2017, 96, e7482. [Google Scholar] [CrossRef]
- Yeremeev, V.; Linge, I.; Kondratieva, T.; Apt, A. Neutrophils exacerbate tuberculosis infection in genetically susceptible mice. Tuberculosis 2015, 95, 447–451. [Google Scholar] [CrossRef]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef]
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A.; Dittel, B.N. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2016, 136, 826–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linnemann, L.C.; Schaible, U.E.; Dallenga, T.K. Evaluation of Myeloperoxidase as Target for Host-Directed Therapy in Tuberculosis In Vivo. Int. J. Mol. Sci. 2022, 23, 2554. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052554
Linnemann LC, Schaible UE, Dallenga TK. Evaluation of Myeloperoxidase as Target for Host-Directed Therapy in Tuberculosis In Vivo. International Journal of Molecular Sciences. 2022; 23(5):2554. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052554
Chicago/Turabian StyleLinnemann, Lara C., Ulrich E. Schaible, and Tobias K. Dallenga. 2022. "Evaluation of Myeloperoxidase as Target for Host-Directed Therapy in Tuberculosis In Vivo" International Journal of Molecular Sciences 23, no. 5: 2554. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052554