
Histone Deacetylase Inhibitors Downregulate Calcium Pyrophosphate Crystal Formation in Human Articular Chondrocytes

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
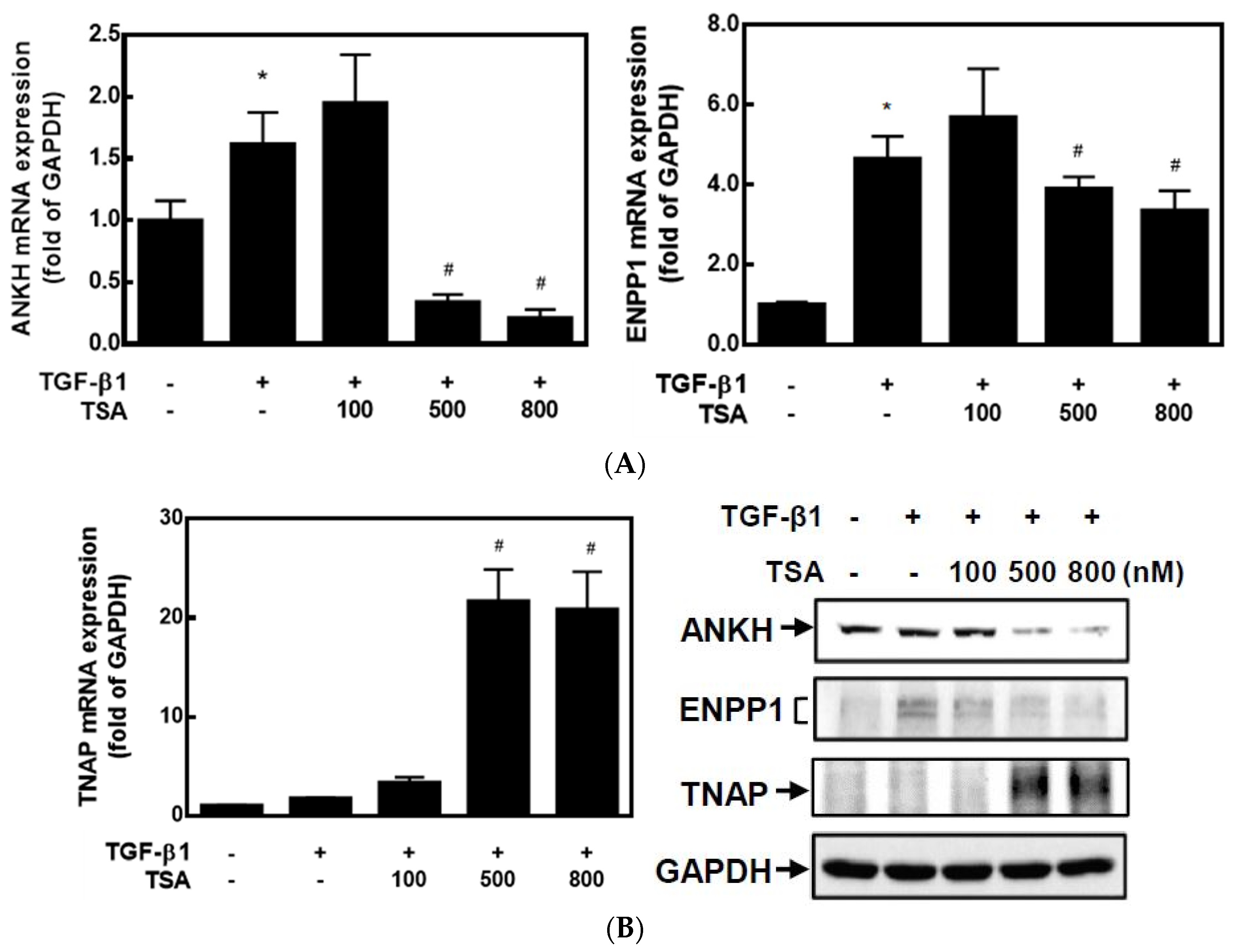
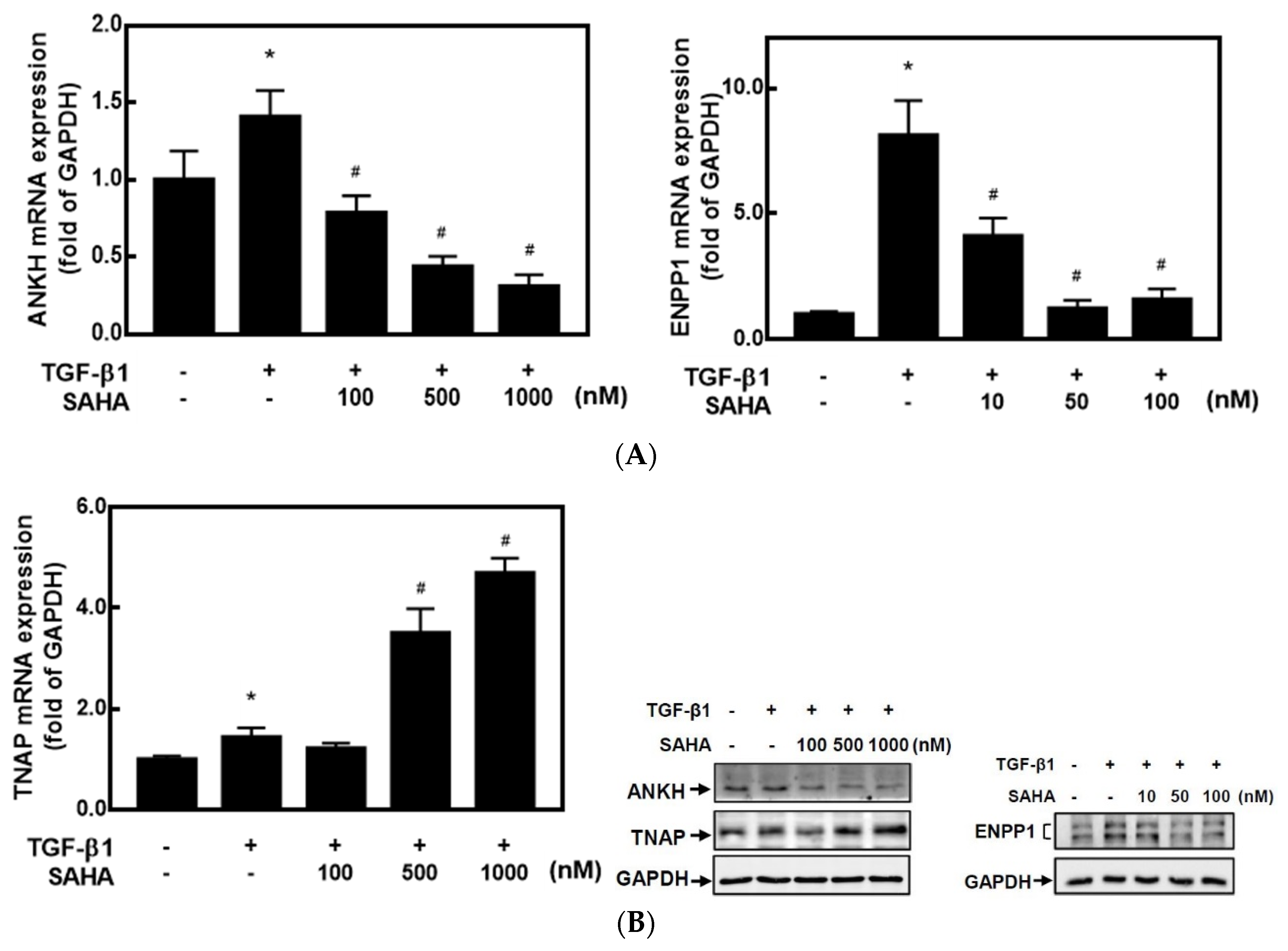
2.1. HDACis Downregulated ANKH and ENPP1 Expressions and Upregulated TNAP Expression
2.2. HDACis Significantly Downregulated ENPP1 and Upregulated TNAP Activities in HC-a Cells
2.3. HDACis Downregulated Extracellular Pyrophosphate Levels and Prevented CPP Crystal Formation
2.4. HDACis Increased the Acetylation of Histones H3 and H4
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Real-Time Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
4.4. Western Blot Analysis
4.5. ENPP1 and TNAP Colorimetric Activity Assays
4.6. Extracellular Pyrophosphate (ePPi) Assay
4.7. CPP Crystal Staining Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCarthy, G.M.; Dunne, A. Calcium crystal deposition diseases—Beyond gout. Nat. Rev. Rheumatol. 2018, 14, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Sidari, A.; Hill, E. Diagnosis and Treatment of Gout and Pseudogout for Everyday Practice. Prim. Care 2018, 45, 213–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Brown, M.A. Genetic studies of chondrocalcinosis. Curr. Opin. Rheumatol. 2005, 17, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Rachow, J.W.; Ryan, L.M. Adenosine triphosphate pyrophosphohydrolase and neutral inorganic pyrophosphatase in pathologic joint fluids. Elevated pyrophosphohydrolase in calcium pyrophosphate dihydrate crystal deposition disease. Arthritis Rheum. 1985, 28, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.K. Crystals, inflammation, and osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 170–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yashiro, T.; Okamoto, T.; Tanaka, R.; Ito, K.; Hara, H.; Yamashita, T.; Kanaji, Y.; Kodama, T.; Ito, Y.; Obara, T.; et al. Prevalence of chondrocalcinosis in patients with primary hyperparathyroidism in Japan. Endocrinol. Jpn. 1991, 38, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Siva, C. Magnesium disorders can cause calcium pyrophosphate deposition disease: A case report and literature review. Eur. J. Rheumatol. 2018, 5, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Mitton-Fitzgerald, E.; Gohr, C.M.; Williams, C.M.; Rosenthal, A.K. Identification of Common Pathogenic Pathways Involved in Hemochromatosis Arthritis and Calcium Pyrophosphate Deposition Disease: A Review. Curr. Rheumatol. Rep. 2022, 24, 40–45. [Google Scholar] [CrossRef]
- Danino, O.; Svetitsky, S.; Kenigsberg, S.; Levin, A.; Journo, S.; Gold, A.; Drexler, M.; Snir, N.; Elkayam, O.; Fischer, B.; et al. Inhibition of nucleotide pyrophosphatase/phosphodiesterase 1: Implications for developing a calcium pyrophosphate deposition disease modifying drug. Rheumatology 2018, 57, 1472–1480. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.; Hashimoto, S.; Lotz, M.; Pritzker, K.; Goding, J.; Terkeltaub, R. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis Rheum. 2001, 44, 1071–1081. [Google Scholar] [CrossRef]
- Williams, C.J. The role of ANKH in pathologic mineralization of cartilage. Curr. Opin. Rheumatol. 2016, 28, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Vollmayer, P.; Clair, T.; Goding, J.W.; Sano, K.; Servos, J.; Zimmermann, H. Hydrolysis of diadenosine polyphosphates by nucleotide pyrophosphatases/phosphodiesterases. Eur. J. Biochem. 2003, 270, 2971–2978. [Google Scholar] [CrossRef]
- Abhishek, A.; Doherty, M. Pathophysiology of articular chondrocalcinosis--role of ANKH. Nat. Rev. Rheumatol. 2011, 7, 96–104. [Google Scholar] [CrossRef]
- Costello, J.C.; Rosenthal, A.K.; Kurup, I.V.; Masuda, I.; Medhora, M.; Ryan, L.M. Parallel regulation of extracellular ATP and inorganic pyrophosphate: Roles of growth factors, transduction modulators, and ANK. Connect. Tissue Res. 2011, 52, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Derfus, B.A.; Camacho, N.P.; Olmez, U.; Kushnaryov, V.M.; Westfall, P.R.; Ryan, L.M.; Rosenthal, A.K. Transforming growth factor beta-1 stimulates articular chondrocyte elaboration of matrix vesicles capable of greater calcium pyrophosphate precipitation. Osteoarthr. Cartil. 2001, 9, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotz, M.; Rosen, F.; McCabe, G.; Quach, J.; Blanco, F.; Dudler, J.; Solan, J.; Goding, J.; Seegmiller, J.E.; Terkeltaub, R. Interleukin 1 beta suppresses transforming growth factor-induced inorganic pyrophosphate (PPi) production and expression of the PPi-generating enzyme PC-1 in human chondrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10364–10368. [Google Scholar] [CrossRef] [Green Version]
- Ryan, L.M.; Rosenthal, A.K. Metabolism of extracellular pyrophosphate. Curr. Opin. Rheumatol. 2003, 15, 311–314. [Google Scholar] [CrossRef]
- Heinkel, D.; Gohr, C.M.; Uzuki, M.; Rosenthal, A.K. Transglutaminase contributes to CPPD crystal formation in osteoarthritis. Front. Biosci. 2004, 9, 3257–3261. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, A.K.; Masuda, I.; Gohr, C.M.; Derfus, B.A.; Le, M. The transglutaminase, Factor XIIIA, is present in articular chondrocytes. Osteoarthr. Cartil. 2001, 9, 578–581. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, A.K.; Gohr, C.M.; Uzuki, M.; Masuda, I. Osteopontin promotes pathologic mineralization in articular cartilage. Matrix Biol. 2007, 26, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Kruhlak, M.J.; Hendzel, M.J.; Fischle, W.; Bertos, N.R.; Hameed, S.; Yang, X.J.; Verdin, E.; Bazett-Jones, D.P. Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J. Biol. Chem. 2001, 276, 38307–38319. [Google Scholar] [CrossRef]
- Buchwald, M.; Krämer, O.H.; Heinzel, T. HDACi—Targets beyond chromatin. Cancer Lett. 2009, 280, 160–167. [Google Scholar] [CrossRef]
- Drysdale, M.J.; Brough, P.A.; Massey, A.; Jensen, M.R.; Schoepfer, J. Targeting Hsp90 for the treatment of cancer. Curr. Opin. Drug Discov. Devel. 2006, 9, 483–495. [Google Scholar] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hammerick, K.E.; James, A.W.; Carre, A.L.; Leucht, P.; Giaccia, A.J.; Longaker, M.T. Inhibition of histone deacetylase activity in reduced oxygen environment enhances the osteogenesis of mouse adipose-derived stromal cells. Tissue Eng. Part A 2009, 15, 3697–3707. [Google Scholar] [CrossRef] [Green Version]
- Destaing, O.; Saltel, F.; Gilquin, B.; Chabadel, A.; Khochbin, S.; Ory, S.; Jurdic, P. A novel Rho-mDia2-HDAC6 pathway controls podosome patterning through microtubule acetylation in osteoclasts. J. Cell Sci. 2005, 118 Pt 13, 2901–2911. [Google Scholar] [CrossRef] [Green Version]
- Huynh, N.C.; Everts, V.; Pavasant, P.; Ampornaramveth, R.S. Inhibition of Histone Deacetylases Enhances the Osteogenic Differentiation of Human Periodontal Ligament Cells. J. Cell Biochem. 2016, 117, 1384–1395. [Google Scholar] [CrossRef]
- Huynh, N.C.; Everts, V.; Nifuji, A.; Pavasant, P.; Ampornaramveth, R.S. Histone deacetylase inhibition enhances in-vivo bone regeneration induced by human periodontal ligament cells. Bone 2017, 95, 76–84. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Liu-Bryan, R.; Pritzker, K.; Firestein, G.S.; Robert Terkeltaub, R. TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J. Immunol. 2005, 174, 5016–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, L.; Hayes, C.P.; Buac, K.; Yoo, D.G.; Rada, B. Pseudogout-associated inflammatory calcium pyrophosphate dihydrate microcrystals induce formation of neutrophil extracellular traps. J. Immunol. 2013, 190, 6488–6500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Doherty, M.; Pascual, E.; Barskova, V.; Guerne, P.A.; Jansen, T.L.; Leeb, B.F.; Perez-Ruiz, F.; Pimentao, J.; Punzi, L.; et al. EULAR recommendations for calcium pyrophosphate deposition. Part II: Management. Ann. Rheum. Dis. 2011, 70, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Ramakrishnan, S.; Mishra, K.; Srivastava, R.; Agarwal, G.G.; Singh, R.; Sircar, A.R. A randomized controlled trial to evaluate the slow-acting symptom-modifying effects of colchicine in osteoarthritis of the knee: A preliminary report. Arthritis Rheum. 2002, 47, 280–284. [Google Scholar] [CrossRef]
- Rothschild, B.; Yakubov, L.E. Prospective 6-month, double-blind trial of hydroxychloroquine treatment of CPDD. Compr. Ther. 1997, 23, 327–331. [Google Scholar]
- Chollet-Janin, A.; Finckh, A.; Dudler, J.; Guerne, P.A. Methotrexate as an alternative therapy for chronic calcium pyrophosphate deposition disease: An exploratory analysis. Arthritis Rheum. 2007, 56, 688–692. [Google Scholar] [CrossRef]
- Cailotto, F.; Bianchi, A.; Sebillaud, S.; Venkatesan, N.; Moulin, D.; Jouzeau, J.Y.; Netter, P. Inorganic pyrophosphate generation by transforming growth factor-beta-1 is mainly dependent on ANK induction by Ras/Raf-1/extracellular signal-regulated kinase pathways in chondrocytes. Arthritis Res. Ther. 2007, 9, R122. [Google Scholar] [CrossRef] [Green Version]
- Rosen, F.; McCabe, G.; Quach, J.; Solan, J.; Terkeltaub, R.; Seegmiller, J.E.; Lotz, M. Differential effects of aging on human chondrocyte responses to transforming growth factor beta: Increased pyrophosphate production and decreased cell proliferation. Arthritis Rheum. 1997, 40, 1275–1281. [Google Scholar]
- Shirakawa, M.; Shiba, H.; Nakanishi, K.; Ogawa, T.; Okamoto, H.; Nakashima, K.; Noshiro, M.; Kato, Y. Transforming growth factor-beta-1 reduces alkaline phosphatase mRNA and activity and stimulates cell proliferation in cultures of human pulp cells. J. Dent. Res. 1994, 73, 1509–1514. [Google Scholar] [CrossRef]
- Guilak, F.; Meyer, B.C.; Ratcliffe, A.; Mow, V.C. The effects of matrix compression on proteoglycan metabolism in articular cartilage explants. Osteoarthr. Cartil. 1994, 2, 91–101. [Google Scholar] [CrossRef]
- Lee, D.A.; Bader, D.L. Compressive strains at physiological frequencies influence the metabolism of chondrocytes seeded in agarose. J. Orthop. Res. 1997, 15, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Graff, R.D.; Lazarowski, E.R.; Banes, A.J.; Lee, G.M. ATP release by mechanically loaded porcine chondrons in pellet culture. Arthritis Rheum. 2000, 43, 1571–1579. [Google Scholar] [CrossRef]
- Xu, H.G.; Hu, C.J.; Wang, H.; Liu, P.; Yang, X.M.; Zhang, Y.; Wang, L.T. Effects of mechanical strain on ANK, ENPP1 and TGF-β1 expression in rat endplate chondrocytes in vitro. Mol. Med. Rep. 2011, 4, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Hirose, J.; Ryan, L.M.; Masuda, I. Up-regulated expression of cartilage intermediate-layer protein and ANK in articular hyaline cartilage from patients with calcium pyrophosphate dihydrate crystal deposition disease. Arthritis Rheum. 2002, 46, 3218–3229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Johnson, K.; Russell, R.G.; Wordsworth, B.P.; Carr, A.J.; Terkeltaub, R.A.; Brown, M.A. Association of sporadic chondrocalcinosis with a -4-basepair G-to-A transition in the 5′-untranslated region of ANKH that promotes enhanced expression of ANKH protein and excess generation of extracellular inorganic pyrophosphate. Arthritis Rheum. 2005, 52, 1110–1117. [Google Scholar] [CrossRef]
- Conway, R.; McCarthy, G.M. Calcium-Containing Crystals and Osteoarthritis: An Unhealthy Alliance. Curr. Rheumatol. Rep. 2018, 20, 13. [Google Scholar] [CrossRef]
- Fuerst, M.; Bertrand, J.; Lammers, L.; Dreier, R.; Echtermeyer, F.; Nitschke, Y.; Rutsch, F.; Schäfer, F.K.; Niggemeyer, O.; Steinhagen, J.; et al. Calcification of articular cartilage in human osteoarthritis. Arthritis Rheum. 2009, 60, 2694–2703. [Google Scholar] [CrossRef]
- Sun, Y.; Mauerhan, D.R.; Honeycutt, P.R.; Kneisl, J.S.; Norton, H.J.; Zinchenko, N.; Hanley, E.N., Jr.; Gruber, H.E. Calcium deposition in osteoarthritic meniscus and meniscal cell culture. Arthritis Res. Ther. 2010, 12, R56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ji, L.; Yang, Y.; Zhang, X.; Gang, Y.; Bai, L. The Role of HDACs and HDACi in Cartilage and Osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 560117. [Google Scholar] [CrossRef]
- Carpio, L.R.; Bradley, E.W.; McGee-Lawrence, M.E.; Weivoda, M.M.; Poston, D.D.; Dudakovic, A.; Xu, M.; Tchkonia, T.; Kirkland, J.L.; van Wijnen, A.J.; et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci. Signal. 2016, 9, ra79. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Derfoul, A.; Pereira-Mouries, L.; Hall, D.J. A novel domain in histone deacetylase 1 and 2 mediates repression of cartilage-specific genes in human chondrocytes. FASEB J. 2009, 23, 3539–3552. [Google Scholar] [CrossRef] [Green Version]
- Chabane, N.; Zayed, N.; Afif, H.; Mfuna-Endam, L.; Benderdour, M.; Boileau, C.; Martel-Pelletier, J.; Pelletier, J.P.; Duval, N.; Fahmi, H. Histone deacetylase inhibitors suppress interleukin-1beta-induced nitric oxide and prostaglandin E2 production in human chondrocytes. Osteoarthr. Cartil. 2008, 16, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Yin, S.; Yang, J.; Jiang, Q.; Cao, W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res. Ther. 2015, 17, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.M.; Ding, Q.H.; Chen, W.P.; Luo, R.B. Vorinostat, a HDAC inhibitor, showed anti-osteoarthritic activities through inhibition of iNOS and MMP expression, p38 and ERK phosphorylation and blocking NF-κB nuclear translocation. Int. Immunopharmacol. 2013, 17, 329–335. [Google Scholar] [CrossRef]
- Vaissière, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef]
- Scott, G.K.; Mattie, M.D.; Berger, C.E.; Benz, S.C.; Benz, C.C. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006, 66, 1277–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, S.; Meng, F.; Wang, J.; Zhang, Y.; Dai, E.; Yu, X.; Li, X.; Jiang, W. SM2miR: A database of the experimentally validated small molecules’ effects on microRNA expression. Bioinformatics 2013, 29, 409–411. [Google Scholar] [CrossRef]
- Suk, F.M.; Chang, C.C.; Lin, R.J.; Lin, S.Y.; Liu, S.C.; Jau, C.F.; Liang, Y.C. ZFP36L1 and ZFP36L2 inhibit cell proliferation in a cyclin D-dependent and p53-independent manner. Sci. Rep. 2018, 8, 2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suk, F.M.; Chang, C.C.; Sun, P.C.; Ke, W.T.; Chung, C.C.; Lee, K.L.; Chan, T.S.; Liang, Y.C. MCPIP1 Enhances TNF-α-Mediated Apoptosis through Downregulation of the NF-κB/cFLIP Axis. Biology 2021, 10, 655. [Google Scholar] [CrossRef]
- Chang, C.C.; Tsai, Y.H.; Liu, Y.; Lin, S.Y.; Liang, Y.C. Calcium-containing crystals enhance receptor activator of nuclear factor κB ligand/macrophage colony-stimulating factor-mediated osteoclastogenesis via extracellular-signal-regulated kinase and p38 pathways. Rheumatology 2015, 54, 1913–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-C.; Lee, K.-L.; Chan, T.-S.; Chung, C.-C.; Liang, Y.-C. Histone Deacetylase Inhibitors Downregulate Calcium Pyrophosphate Crystal Formation in Human Articular Chondrocytes. Int. J. Mol. Sci. 2022, 23, 2604. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052604
Chang C-C, Lee K-L, Chan T-S, Chung C-C, Liang Y-C. Histone Deacetylase Inhibitors Downregulate Calcium Pyrophosphate Crystal Formation in Human Articular Chondrocytes. International Journal of Molecular Sciences. 2022; 23(5):2604. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052604
Chicago/Turabian StyleChang, Chi-Ching, Kun-Lin Lee, Tze-Sian Chan, Chia-Chen Chung, and Yu-Chih Liang. 2022. "Histone Deacetylase Inhibitors Downregulate Calcium Pyrophosphate Crystal Formation in Human Articular Chondrocytes" International Journal of Molecular Sciences 23, no. 5: 2604. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052604