
Analytical Assessment of the Vela Diagnostics NGS Assay for HIV Genotyping and Resistance Testing: The Apulian Experience

 ,
,
Abstract
:
1. Introduction
2. Results
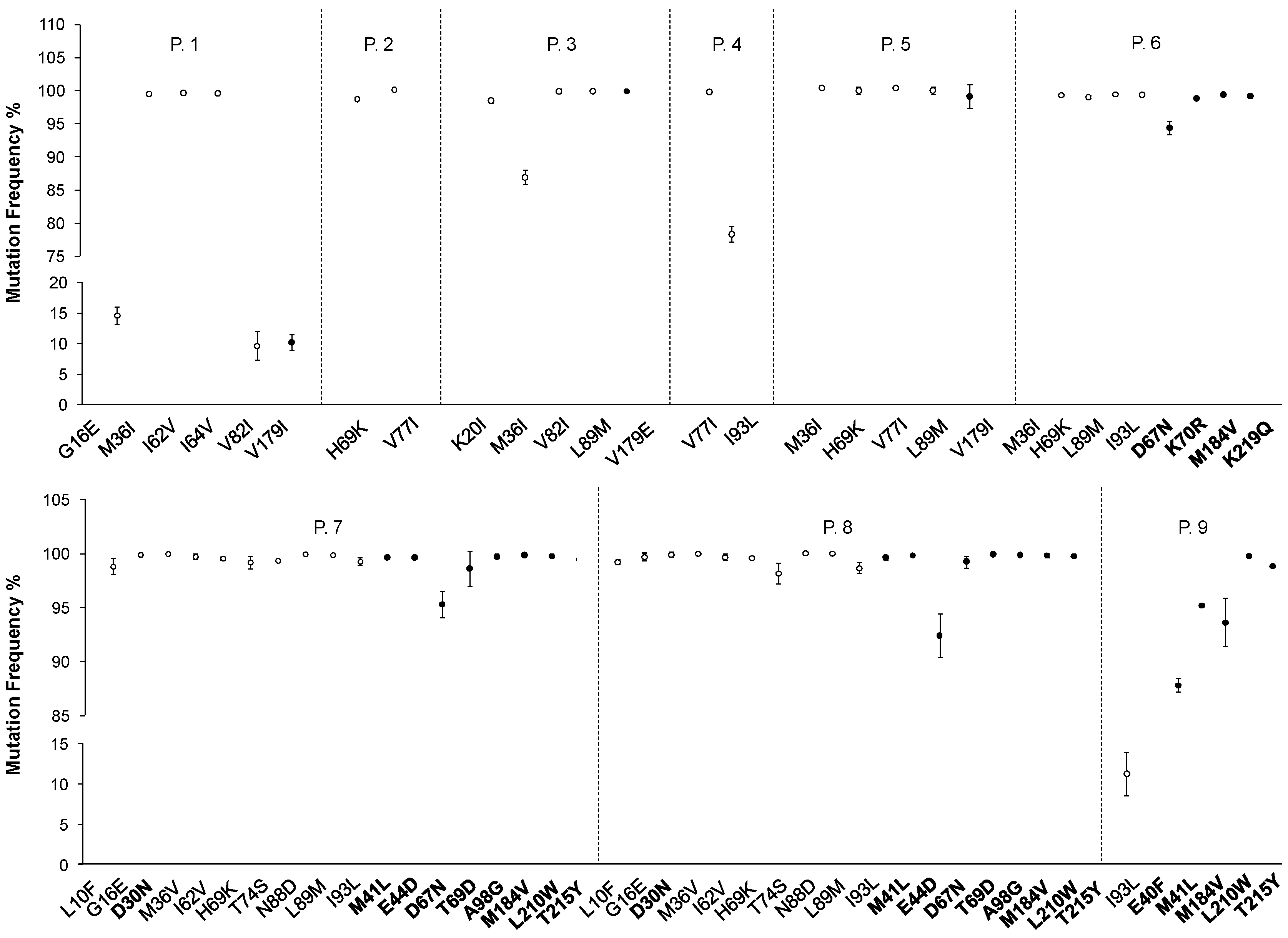
2.1. NGS Analyses Performed on Clinical Samples
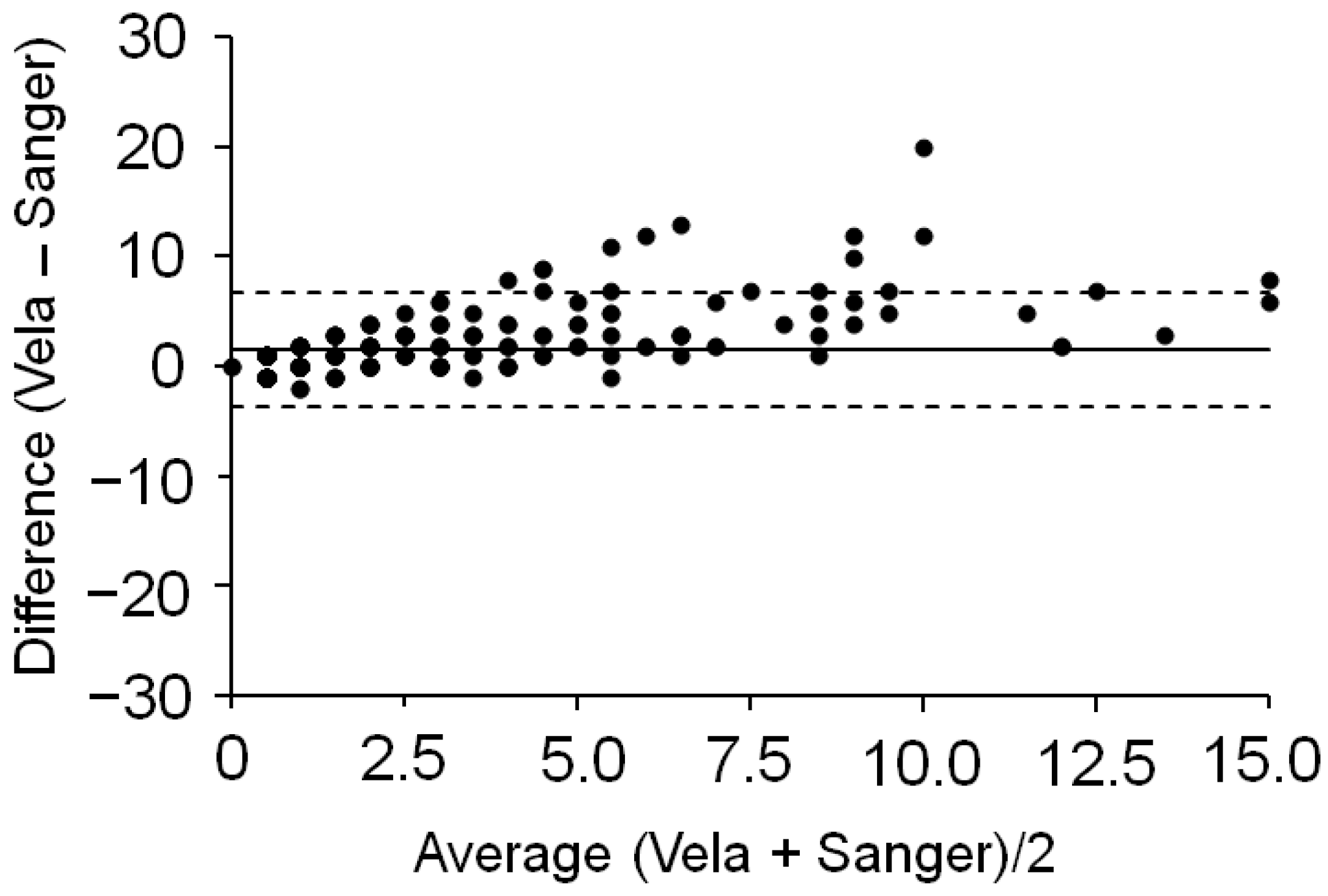
2.2. Comparison with Sanger Sequencing
2.3. NGS Sequencing of Reference Samples
2.3.1. Accuracy Performance of the Vela Diagnostics NGS Platform
2.3.2. Intra-Assay Repeatability
2.3.3. Inter-Assay Reproducibility
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Data Analysis
4.2. Next-Generation Sequencing with the Vela Diagnostics Platform
4.3. Dideoxynucleoside Sanger Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deeks, S.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 10 January 2022).
- Gutiérrez-Sevilla, J.E.; Cárdenas-Bedoya, J.; Escoto-Delgadillo, M.; Zúñiga-González, G.M.; Pérez-Ríos, A.M.; Gómez-Meda, B.C.; González-Enríquez, G.C.; Figarola-Centurión, I.; Chavarría-Avila, E.; Torres-Mendoza, B.M. Genomic instability in people living with HIV. Mutat. Res./Genet. Toxicol. Environ. Mutagenes. 2021, 865, 503336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, Q.; Ni, M.; Liu, T.; Wang, C.; Song, C.; Liao, L.; Xing, H.; Jiang, S.; Shao, Y.; et al. Dynamics of HIV-1 quasispecies diversity of participants on long-term antiretroviral therapy based on intrahost single-nucleotide variations. Int. J. Infect. Dis. 2021, 104, 306–314. [Google Scholar] [CrossRef]
- Gianella, S.; Richman, D.D. Minority Variants of Drug-Resistant HIV. J. Infect. Dis. 2010, 202, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Z.; Kuritzkes, D.R. Clinical Implications of HIV-1 Minority Variants. Clin. Infect. Dis. 2013, 56, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenhende, M.-A.; Bellecave, P.; Recordon-Pinson, P.; Reigadas, S.; Bidet, Y.; Bruyand, M.; Bonnet, F.; Lazaro, E.; Neau, D.; Fleury, H.; et al. Prevalence and Evolution of Low Frequency HIV Drug Resistance Mutations Detected by Ultra Deep Sequencing in Patients Experiencing First Line Antiretroviral Therapy Failure. PLoS ONE 2014, 9, e86771. [Google Scholar] [CrossRef]
- Halvas, E.K.; Wiegand, A.; Boltz, V.F.; Kearney, M.; Nissley, D.; Wantman, M.; Hammer, S.M.; Palmer, S.; Vaida, F.; Coffin, J.M.; et al. Low Frequency Nonnucleoside Reverse-Transcriptase Inhibitor–Resistant Variants Contribute to Failure of Efavirenz-Containing Regimens in Treatment-Experienced Patients. J. Infect. Dis. 2010, 201, 672–680. [Google Scholar] [CrossRef]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar]
- World Health Organization. Guidelines on the Public Health Response to Pretreatment HIV Drug Resistance; WHO: Geneva, Switzerland, 2017; ISBN 978-92-4-155005-5. [Google Scholar]
- Saag, M.S.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Thompson, M.A.; Sax, P.E.; Smith, D.M.; Benson, C.A.; Buchbinder, S.P.; del Rio, C.; et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2020 recommendations of the International Antiviral Society-USA Panel. JAMA 2020, 324, 1651–1669. [Google Scholar] [CrossRef]
- Lee, E.R.; Gao, F.; Sandstrom, P.; Ji, H. External Quality Assessment for Next-Generation Sequencing-Based HIV Drug Resistance Testing: Unique Requirements and Challenges. Viruses 2020, 12, 550. [Google Scholar] [CrossRef]
- Stella-Ascariz, N.; Arribas, J.R.; Paredes, R.; Li, J.Z. The role of HIV-1 drug-resistant minority variants in treatment failure. J. Infect. Dis. 2017, 216, S847–S850. [Google Scholar] [CrossRef] [PubMed]
- May, S.; Adamska, E.; Tang, J. Evaluation of Vela Diagnostics HIV-1 genotyping assay on an automated next generation sequencing platform. J. Clin. Virol. 2020, 127, 104376. [Google Scholar] [CrossRef] [PubMed]
- Gholami, M.; Rouzbahani, N.; Samiee, S.; Tayeri, K.; Ghorban, K.; Dehkharghani, A.D.; Gholami, A.A.; Moshiri, F.; Sattari, A.; Dadmanesh, M.; et al. HIV-1 drug resistance mutations detection and HIV-1 subtype G report by using next-generation sequencing platform. Microb. Pathog. 2020, 146, 104221. [Google Scholar] [CrossRef] [PubMed]
- Raymond, S.; Nicot, F.; Abravanel, F.; Minier, L.; Carcenac, R.; Lefebvre, C.; Harter, A.; Martin-Blondel, G.; Delobel, P.; Izopet, J. Performance evaluation of the Vela Dx Sentosa next-generation sequencing system for HIV-1 DNA genotypic resistance. J. Clin. Virol. 2020, 122, 104229. [Google Scholar] [CrossRef]
- Sulis, G.; The HIV/Migrants Study Group; El Hamad, I.; Fabiani, M.; Rusconi, S.; Maggiolo, F.; Guaraldi, G.; Bozzi, G.; Bernardini, C.; Lichtner, M.; et al. Clinical and epidemiological features of HIV/AIDS infection among migrants at first access to healthcare services as compared to Italian patients in Italy: A retrospective multicentre study, 2000–2010. Infection 2014, 42, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Simonetti, F.R.; Brindicci, G.; Bergna, A.; Di Giambenedetto, S.; Sterrantino, G.; Mussini, C.; Menzo, S.; Bagnarelli, P.; Zazzi, M.; et al. Local epidemics gone viral: Evolution and diffusion of the Italian HIV-1 recombinant form CRF60_BC. Front. Microbiol. 2019, 10, 769. [Google Scholar] [CrossRef]
- Rhee, S.-Y.; Kantor, R.; Katzenstein, D.; Camacho, R.; Morris, L.; Sirivichayakul, S.; Jorgensen, L.; Brigido, L.; Schapiro, J.M.; Shafer, R.W. HIV-1 pol mutation frequency by subtype and treatment experience: Extension of the HIVseq program to seven non-B subtypes. AIDS 2006, 20, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.L.; Scheier, T.; Ledermann, U.; Flepp, M.; Metzner, K.J.; Böni, J.; Günthard, H.F. Emergence of Resistance to Integrase Strand Transfer Inhibitors during Dolutegravir Containing Triple-Therapy in a Treatment-Experienced Patient with Pre-Existing M184V/I Mutation. Viruses 2020, 12, 1330. [Google Scholar] [CrossRef]
- Chaplin, B.; Imade, G.; Onwuamah, C.; Odaibo, G.; Audu, R.; Okpokwu, J.; Olaleye, D.; Meloni, S.; Rawizza, H.; Muazu, M.; et al. Distinct Pattern of Thymidine Analogue Mutations with K65R in Patients Failing Tenofovir-Based Antiretroviral Therapy. AIDS Res. Hum. Retrovir. 2018, 34, 228–233. [Google Scholar] [CrossRef]
- Penrose, K.J.; Wallis, C.; Brumme, C.J.; Hamanishi, K.A.; Gordon, K.C.; Viana, R.V.; Harrigan, P.R.; Mellors, J.W.; Parikh, U.M. Frequent Cross-Resistance to Dapivirine in HIV-1 Subtype C-Infected Individuals after First-Line Antiretroviral Therapy Failure in South Africa. Antimicrob. Agents Chemother. 2017, 61, e01805-16. [Google Scholar] [CrossRef] [Green Version]
- Omooja, J.; Nannyonjo, M.; Sanyu, G.; Nabirye, S.E.; Nassolo, F.; Lunkuse, S.; Kapaata, A.; Segujja, F.; Kateete, D.P.; Ssebaggala, E.; et al. Rates of HIV-1 virological suppression and patterns of acquired drug resistance among fisherfolk on first-line antiretroviral therapy in Uganda. J. Antimicrob. Chemother. 2019, 74, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Parkin, N.T.; Gupta, S.; Chappey, C.; Petropoulos, C.J. The K101P and K103R/V179D Mutations in Human Immunodeficiency Virus Type 1 Reverse Transcriptase Confer Resistance to Nonnucleoside Reverse Transcriptase Inhibitors. Antimicrob. Agents Chemother. 2006, 50, 351–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Odoom, A.; Brown, C.A.; Odoom, J.K.; Bonney, E.Y.; Ntim, N.A.A.; Delgado, E.; Lartey, M.; Sagoe, K.W.; Adiku, T.; Ampofo, W.K. Emergence of HIV-1 drug resistance mutations in mothers on treatment with a history of prophylaxis in Ghana. Virol. J. 2018, 15, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluis-Cremer, N. The Emerging Profile of Cross-Resistance among the Nonnucleoside HIV-1 Reverse Transcriptase Inhibitors. Viruses 2014, 6, 2960–2973. [Google Scholar] [CrossRef]
- Xu, H.-T.; Colby-Germinario, S.P.; Asahchop, E.L.; Oliveira, M.; McCallum, M.; Schader, S.M.; Han, Y.; Quan, Y.; Sarafianos, S.G.; Wainberg, M.A. Effect of Mutations at Position E138 in HIV-1 Reverse Transcriptase and Their Interactions with the M184I Mutation on Defining Patterns of Resistance to Nonnucleoside Reverse Transcriptase Inhibitors Rilpivirine and Etravirine. Antimicrob. Agents Chemother. 2013, 57, 3100–3109. [Google Scholar] [CrossRef] [Green Version]
- Raymond, S.; Nicot, F.; Carcenac, R.; Lefebvre, C.; Jeanne, N.; Saune, K.; Delobel, P.; Izopet, J. HIV-1 genotypic resistance testing using the Vela automated next-generation sequencing platform. J. Antimicrob. Chemother. 2018, 73, 1152–1157. [Google Scholar] [CrossRef]
- Liu, T.F.; Shafer, R.W. Web Resources for HIV Type 1 Genotypic-Resistance Test Interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef] [Green Version]
- Tzou, P.L.; Ariyaratne, P.; Varghese, V.; Lee, C.; Rakhmanaliev, E.; Villy, C.; Yee, M.; Tan, K.; Michel, G.; Pinsky, B.A.; et al. Comparison of an In Vitro Diagnostic Next-Generation Sequencing Assay with Sanger Sequencing for HIV-1 Genotypic Resistance Testing. J. Clin. Microbiol. 2018, 56, e00105-18. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.; Volkova, I.; Sahoo, M.K.; Tzou, P.L.; Shafer, R.W.; Pinsky, B.A. Prospective Evaluation of the Vela Diagnostics Next-Generation Sequencing Platform for HIV-1 Genotypic Resistance Testing. J. Mol. Diagn. 2019, 21, 961–970. [Google Scholar] [CrossRef]
- Alidjinou, E.K.; Coulon, P.; Hallaert, C.; Robineau, O.; Meybeck, A.; Huleux, T.; Ajana, F.; Hober, D.; Bocket, L. Routine drug resistance testing in HIV-1 proviral DNA, using an automated next- generation sequencing assay. J. Clin. Virol. 2019, 121, 104207. [Google Scholar] [CrossRef]
- Lapointe, H.R.; Dong, W.; Lee, G.Q.; Bangsberg, D.R.; Martin, J.N.; Mocello, A.R.; Boum, Y.; Karakas, A.; Kirkby, D.; Poon, A.F.Y.; et al. HIV Drug Resistance Testing by High-Multiplex “Wide” Sequencing on the MiSeq Instrument. Antimicrob. Agents Chemother. 2015, 59, 6824–6833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessilly, G.; Goeminne, L.; Vandenbroucke, A.-T.; Dufrasne, F.E.; Martin, A.; Kabamba-Mukabi, B. First evaluation of the Next-Generation Sequencing platform for the detection of HIV-1 drug resistance mutations in Belgium. PLoS ONE 2018, 13, e0209561. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Metzner, K.J.; Geissberger, F.D.; Shah, C.; Leemann, C.; Klimkait, T.; Böni, J.; Trkola, A.; Zagordi, O. MinVar: A rapid and versatile tool for HIV-1 drug resistance genotyping by deep sequencing. J. Virol. Methods 2017, 240, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Sandstrom, P.; Paredes, R.; Harrigan, P.R.; Brumme, C.J.; Rios, S.A.; Noguera-Julian, M.; Parkin, N.; Kantor, R. Are We Ready for NGS HIV Drug Resistance Testing? The Second “Winnipeg Consensus” Symposium. Viruses 2020, 12, 586. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Nomenclature for incompletely specified bases in nucleic acid sequences: Recommendations 1984. Nucleic Acids Res. 1985, 13, 3021–3030. [Google Scholar] [CrossRef]
- Armenia, D.; Fabeni, L.; Alteri, C.; Di Pinto, D.; Di Carlo, D.; Bertoli, A.; Gori, C.; Carta, S.; Fedele, V.; Forbici, F.; et al. HIV-1 integrase genotyping is reliable and reproducible for routine clinical detection of integrase resistance mutations even in patients with low-level viraemia. J. Antimicrob. Chemother. 2015, 70, 1865–1873. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
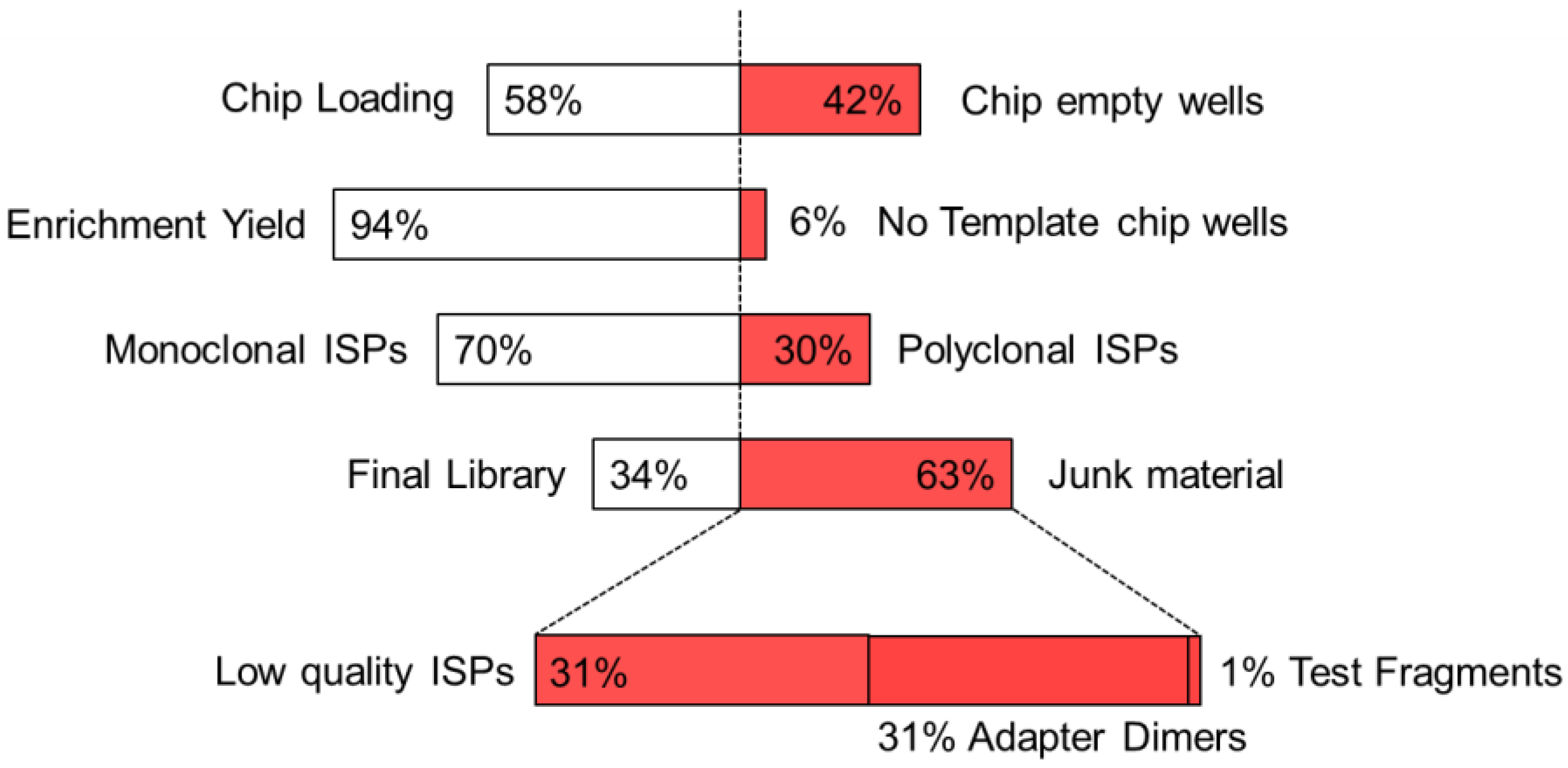{kind=link}
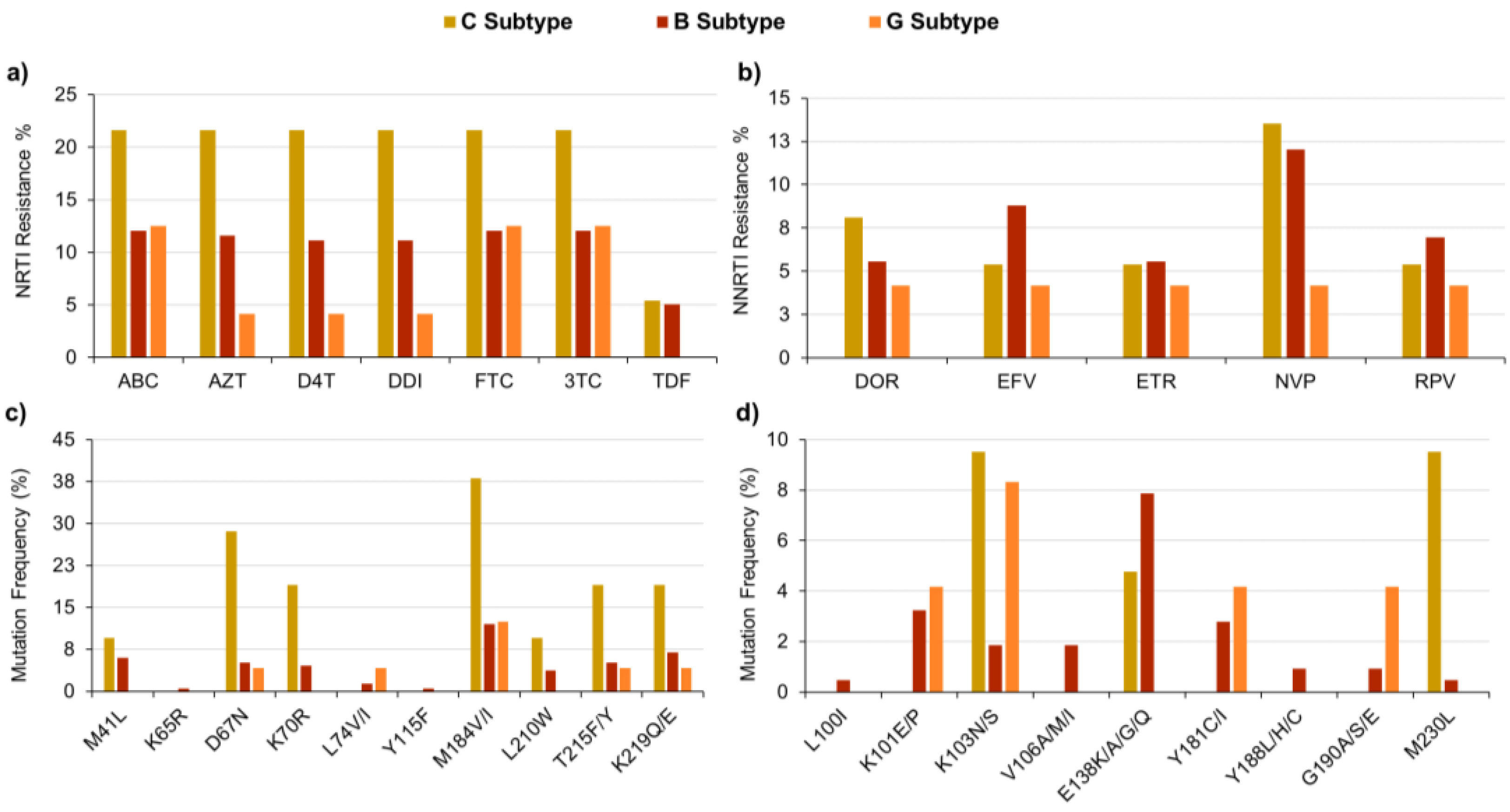{kind=link}
{kind=link}
{kind=link}
| Parameter | Median (IQR) | Percentage |
|---|---|---|
| Sex (male %) | 71.4 | |
| Age (years) | 45 (35–53) | |
| HIV-1 viral load (cp/mL) | 17744 (327–97859) | |
| HIV-1 subtype (B %) | 65.3 | |
| CD45 cell count (cells/µL) | 1960 (1336–2471) | |
| CD4 cell count (cells/µL) | 435 (218–698) | |
| CD4/CD8 ratio | 0.46 (0.22–0.73) |
| NRTI | |||||||
| ABC | AZT | D4T | DDI | FTC | 3TC | TDF | |
| SS | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| NGS | 3 | 3 | 1 | 3 | 5 | 5 | 1 |
| NNRTI | |||||||
| DOR | EFV | ETR | NVP | RPV | |||
| SS | 1 | 1 | 1 | 1 | 1 | ||
| NGS | 3 | 3 | 3 | 4 | 4 | ||
| INSTI | |||||||
| BIC | CAB | DTG | EVG | RAL | |||
| SS | 1 | 1 | 1 | 1 | 1 | ||
| NGS | 2 | 2 | 2 | 3 | 3 | ||
| PRO/RT | Sample 1 | Sample 2 | Sample 3 | Sample 4 | Sample 5 |
|---|---|---|---|---|---|
| Subtype | CRF_02AG | C | B | D | C |
| Median coverage | 10997 | 3134 | 6771 | 9912 | 8694 |
| Total Mapped Reads | 166413 (99.43%) | 39642 (99.14%) | 124209 (99.69%) | 174423 (99.53%) | 156676 (99.54%) |
| Mean Read Length | 153 | 159 | 136 | 146 | 137 |
| Completeness | 94.69% | 94.75% | 94.69% | 94.82% | 94.75 |
| GC content (%) | 38.51% | 38.17% | 38.04% | 37.80% | 39.10% |
| Expected variants | RT: V179I PRO: K20I, M36I, L63S, H69K, L89M | RT: A98G, K103N, M184V, T215Y, M230L PRO: M36I, L63A, H69K, I93L | RT: K103KQ PRO: I64V, V77I, I93L | RT: - PRO: M36I, D60E, I64V | RT: D67N, K70R, M184V, K219Q PRO: M36I, H69K, L89M, I93L |
| Unexpected variants | - | - | - | PRO: A71AT | - |
| INT | Sample 1 | Sample 2 | Sample 3 | Sample 4 | Sample 5 |
|---|---|---|---|---|---|
| Subtype | CRF_02AG | C | B | D | C |
| Median coverage | 3597 | 9127 | 8057 | 8038 | 7665 |
| Total Mapped Reads | 70,284 (99.23%) | 180,631 (99.54%) | 157,328 (99.69%) | 164,132 (99.73%) | 143,912 (99.5%) |
| Read Length | 159 | 153 | 158 | 139 | 144 |
| Completeness | 98.64% | 98.64% | 98.64 | 98.64% | 98.64% |
| GC content (%) | 38.61% | 38.69% | 38.05% | 37.80% | 38.95% |
| Expected variants | T125A, G163E | T125P | - | T125TA | T125A |
| Unexpected variants | - | - | T125TM | T125A | - |
| Sample | Viral Load (cp/mL) | Subtype | Mapped Reads | Coverage | No. Mismatches |
|---|---|---|---|---|---|
| P. 1_A | 928,859 | B | 158218 (99.46%) | PRO/RT: 7543 INT: 7743 | 0 |
| P. 1_B | 130509 (99.46%) | PRO/RT: 9511 INT: 3640 | |||
| P. 2_A | 66,857 | B | 69321 (99.48%) | PRO/RT: 4956 INT: 2629 | 2 |
| P. 2_B | 38263 (99.48%) | PRO/RT: 3165 a INT: 1570 | |||
| P. 3_A | 32,500 | G | 117138 (99.29%) | PRO/RT: 9553 INT: 4119 | 0 |
| P. 3_B | 174204 (99.38%) | PRO/RT: 13,870 INT: 6268 | |||
| P. 4_A | 14,900 | B | 121373 (99.47%) | PRO/RT: 8116 INT: 3746 | 0 |
| P. 4_B | 111858 (99.0%) | PRO/RT: 7262 INT: 3689 | |||
| P. 5_A | 737 | A1 | 33565 (98.92%) | PRO/RT: 106 b INT: 1697 | 1 |
| P. 5_B | 36386 (99.2%) | PRO/RT: 107 INT: 1774 | |||
| P. 6_A | 29,512 | C | 104346 (99.34%) | PRO/RT: 6518 INT: 5266 | 0 |
| P. 6_B | 77317 (99.61%) | PRO/RT: 4658 INT: 4034 | |||
| P. 7_A | 37,153 | C | 41770 (99.45%) | PRO/RT: 2920 INT: 2188 | 0 |
| P. 7_B | 29267 (99.34%) | PRO/RT: 2340 INT: 966 | |||
| P. 8_A | 112,202 | C | 64745 (99.66%) | PRO/RT: 4001 INT: 3925 | 0 |
| P. 8_B | 68129 (99.43%) | PRO/RT: 5364 INT: 3591 | |||
| P. 9_A | 144,544 | B | 132430 (99.59%) | PRO/RT: 10,017 INT: 6036 | 0 |
| P. 9_B | 79885 (99.75%) | PRO/RT: 4584 INT: 5027 |
| Sample | Viral Load (cp/mL) | Subtype | Mapped Reads | Coverage | No. Mismatches |
|---|---|---|---|---|---|
| P. 6_Run A | 423,608 | G | 214,315 (99.23%) | PRO/RT: 15,033 INT: 8023 | 0 |
| P. 6_Run B | 215,497 (99.39%) | PRO/RT: 15,416 INT: 8280 | |||
| * P. 7_Run A | 173,785 | B | 19,908 (98.29%) | PRO/RT: 910 INT: 901 | 0 |
| * P. 7_Run B | 20,764 (98.68%) | PRO/RT: 970 INT: 925 | |||
| P. 8_Run A | 149,917 | B | 138,132 (99.41%) | PRO/RT: 8734 INT: 8051 | 0 |
| P. 8_Run B | 141,556 (99.54%) | PRO/RT: 9072 INT: 8567 | |||
| P. 9_Run A | 129,202 | A1 | 62,630 (98.69%) | PRO/RT: 3995 INT: 3091 | 0 |
| P. 9_Run B | 63,129 (98.87%) | PRO/RT: 4025 INT: 3145 | |||
| P. 10_Run A | 63,341 | B | 161,624 (99.06%) | PRO/RT: 9508 INT: 8339 | 1 |
| P. 10_Run B | 160,832 (99.19%) | PRO/RT: 9567 INT: 8635 | |||
| * P. 11_Run A | 56,746 | B | 1974 (96.67%) | PRO/RT: 121 a INT: 58 | 0 |
| * P. 11_Run B | 50,592 (99.37%) | PRO/RT: 1866 INT: 4057 | |||
| * P. 12_Run A | 25,387 | C | 8102 (98.55%) | PRO/RT: 623 INT: 304 | 0 |
| * P. 12_Run B | 8053 (99.0%) | PRO/RT: 649 INT: 319 | |||
| P. 13_Run A | 6590 | B | 135,416 (99.49%) | PRO/RT: 7207 INT: 6910 | 0 |
| P. 13_Run B | 134,018 (99.58%) | PRO/RT: 7136 INT: 6963 | |||
| * P. 14_Run A | 5953 | A1 | 9707 (99.18%) | PRO/RT: 713 INT: 267 | 3 |
| * P. 14_Run B | 9193 (99.34%) | PRO/RT: 712 INT: 259 | |||
| * P. 15_Run A | 936 | B | 13,183 (99.07%) | PRO/RT: 1093 INT: 302 | 0 |
| * P. 15_Run B | 13,279 (99.25%) | PRO/RT: 1091 INT: 326 | |||
| * P. 16_Run A | 922 | CRF_02AG | 2013 (97.62%) | PRO/RT: 85 b INT: 25 | 1 |
| * P. 16_Run B | 2004 (98.09%) | PRO/RT: 89 c INT: 28 | |||
| * P. 17_Run A | 489 | A1 | 5583 (98.24%) | PRO/RT: 176 INT: 230 | 0 |
| * P. 17_Run B | 5384 (98.81%) | PRO/RT: 156 INT: 224 | |||
| P. 18_Run A | 480 | B | 136,968 (99.2%) | PRO/RT: 4129 INT: 4848 | 0 |
| P. 18_Run B | 135,374 (99.35%) | PRO/RT: 4105 INT: 4854 | |||
| P. 19_Run A | 319 | B | 45,798 (99.78%) | PRO/RT: 1936 INT: NA | NA |
| P. 19_Run B | 19,752 (99.26%) | PRO/RT: NA INT: 339 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonifacio, M.A.; Genchi, C.; Lagioia, A.; Talamo, V.; Volpe, A.; Mariggiò, M.A. Analytical Assessment of the Vela Diagnostics NGS Assay for HIV Genotyping and Resistance Testing: The Apulian Experience. Int. J. Mol. Sci. 2022, 23, 2727. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052727
Bonifacio MA, Genchi C, Lagioia A, Talamo V, Volpe A, Mariggiò MA. Analytical Assessment of the Vela Diagnostics NGS Assay for HIV Genotyping and Resistance Testing: The Apulian Experience. International Journal of Molecular Sciences. 2022; 23(5):2727. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052727
Chicago/Turabian StyleBonifacio, Maria Addolorata, Chiara Genchi, Antonella Lagioia, Vincenza Talamo, Anna Volpe, and Maria Addolorata Mariggiò. 2022. "Analytical Assessment of the Vela Diagnostics NGS Assay for HIV Genotyping and Resistance Testing: The Apulian Experience" International Journal of Molecular Sciences 23, no. 5: 2727. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052727