
Exploring Pharmacological Functions of Alternatively Spliced Variants of the Mu Opioid Receptor Gene, Oprm1, via Gene-Targeted Animal Models

 ,
,
Abstract
:1. Introduction
2. Pharmacology and Molecular Biology of Mu Opioid Receptors
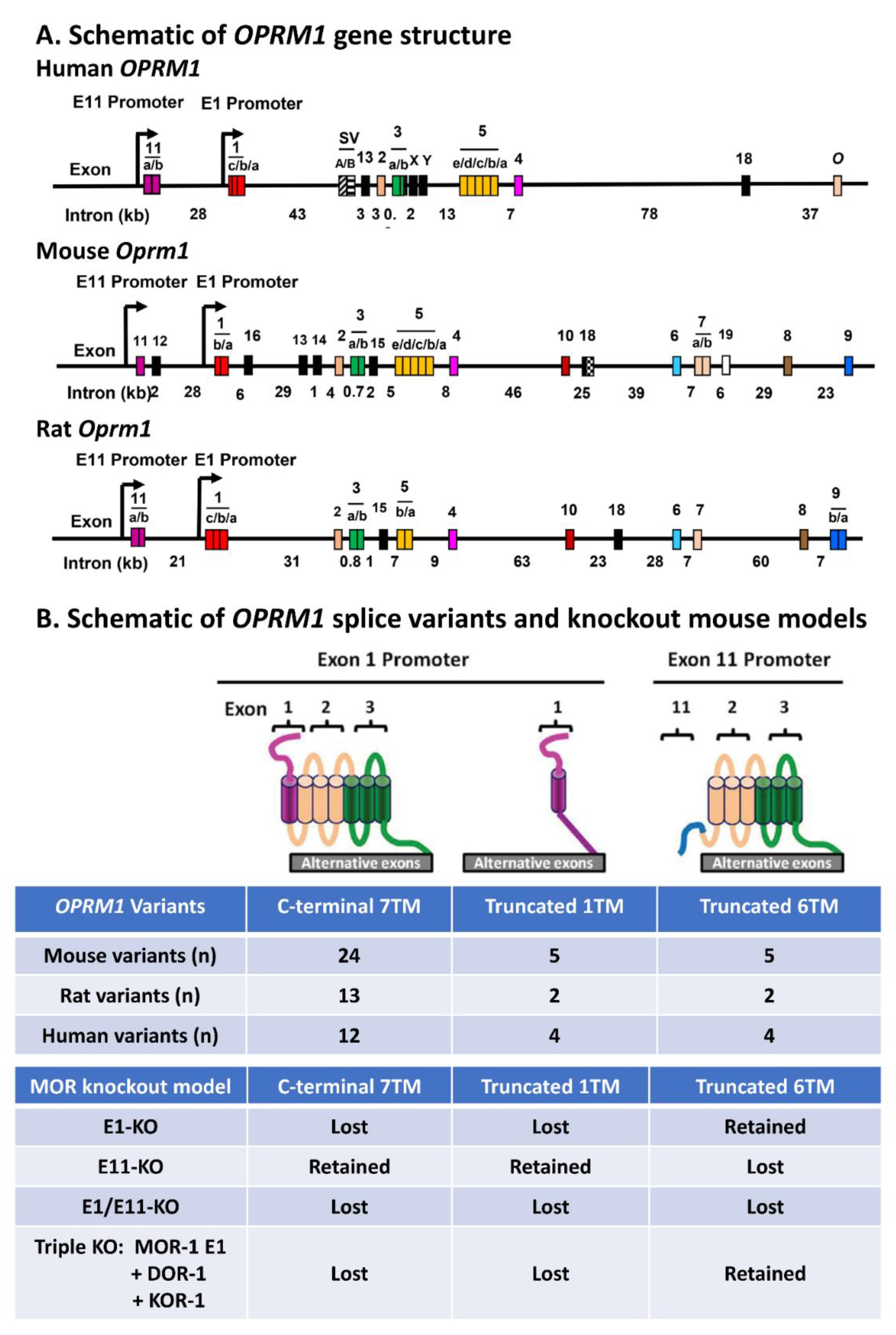
3. Alternative Pre-mRNA Splicing of the Mu Opioid Receptor Gene, OPRM1
4. Targeting Oprm1 in Mice
5. In Vivo Functions of Oprm1 Alternatively Spliced Variants Using Oprm1-Targeted Mouse Models
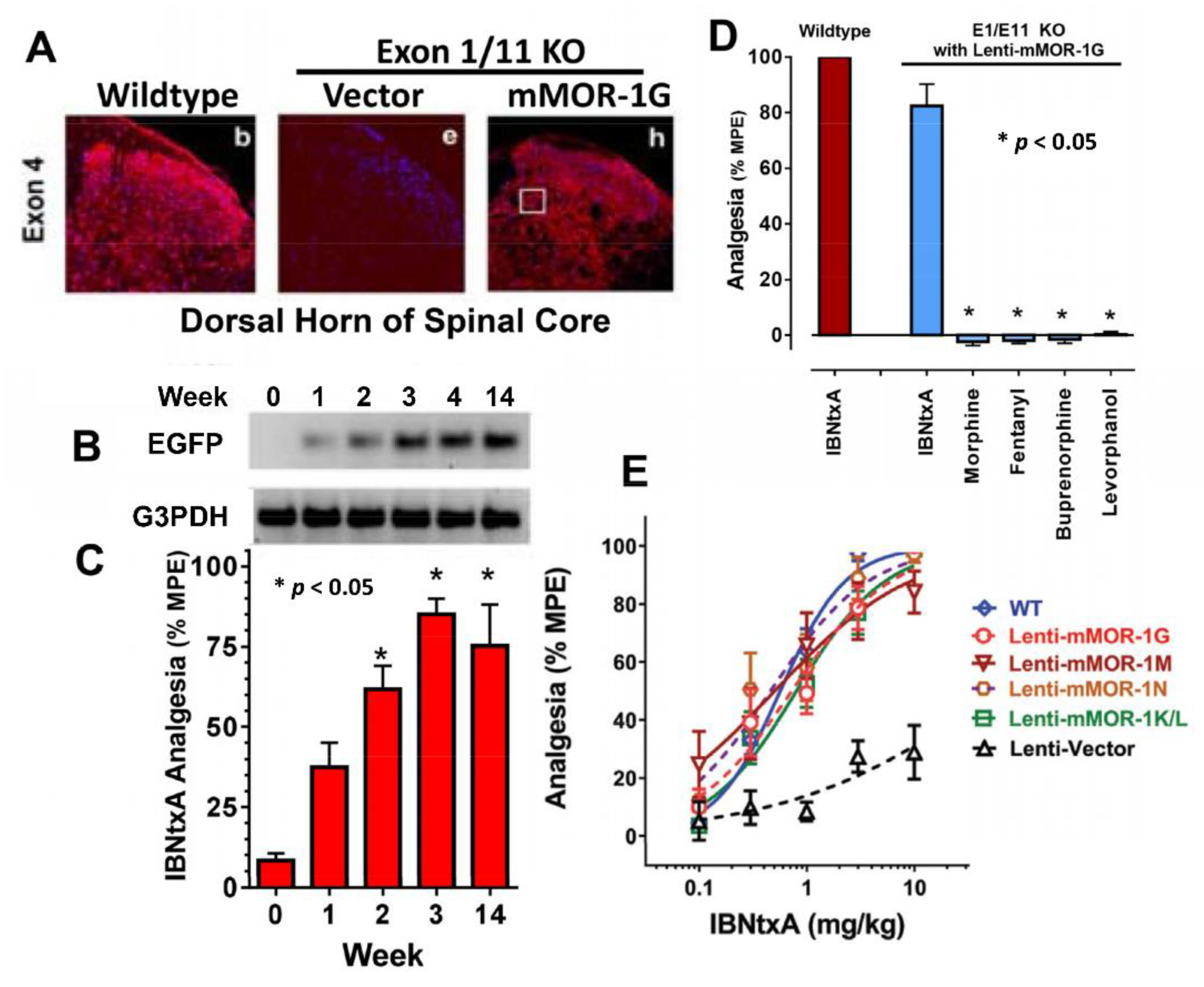
5.1. In Vivo Functions of Exon 11-Associated Oprm1 6TM Variants: Targeting Exons 1 and 11
5.1.1. The Role of 6TM Variants in M6G and Heroin Analgesia
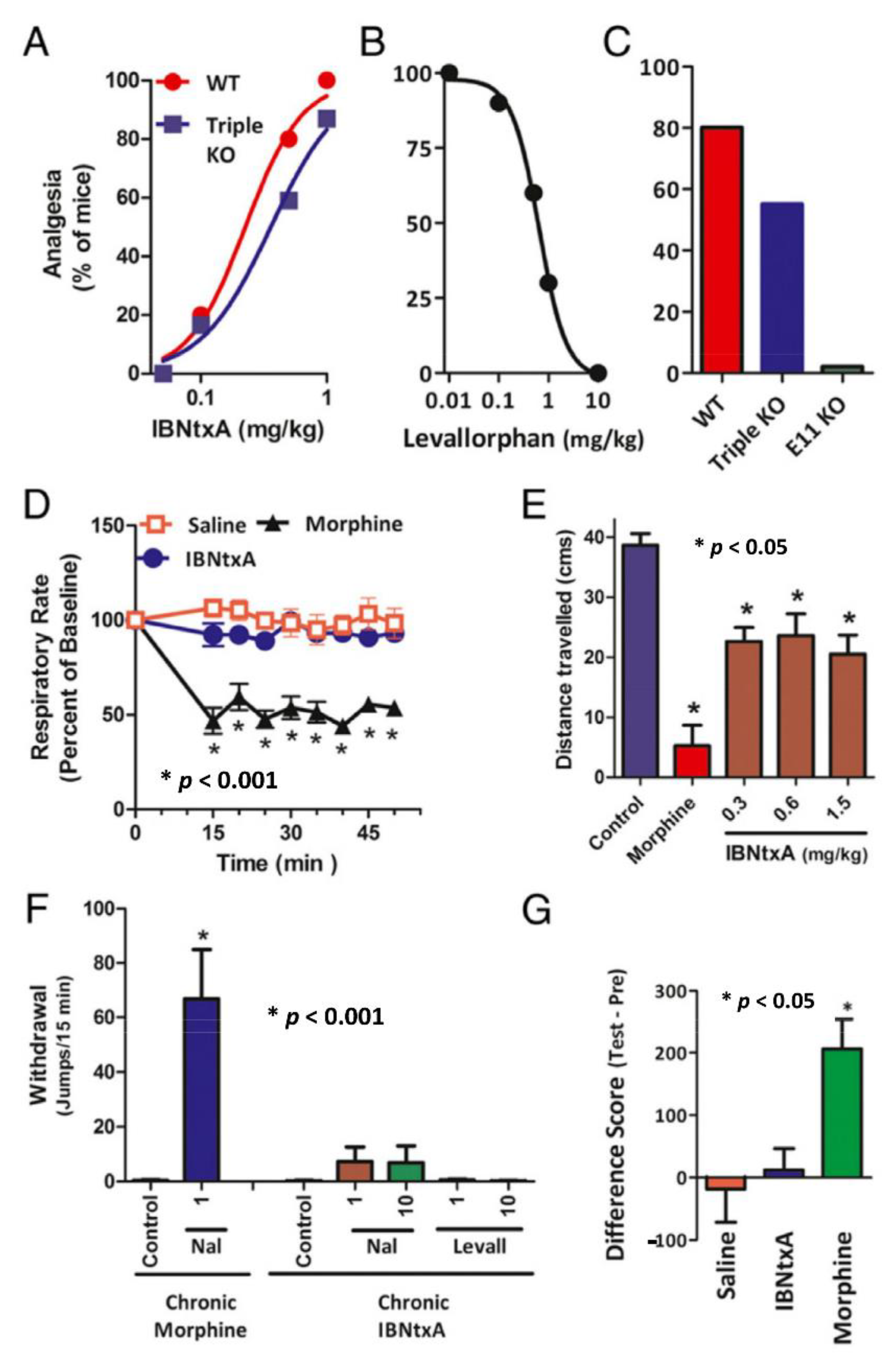
5.1.2. The Role of 6TM Variants in the Analgesic Action of Arylepoxamides, a New Class of Potent and Safer Analgesics
5.1.3. Classification of Mu Opioids Based on Alternatively Spliced Oprm1 Variants
5.1.4. The Role of 6TM Variants in the Analgesic Action of Delta Opioids, Kappa Opioids, and Non-Opioids
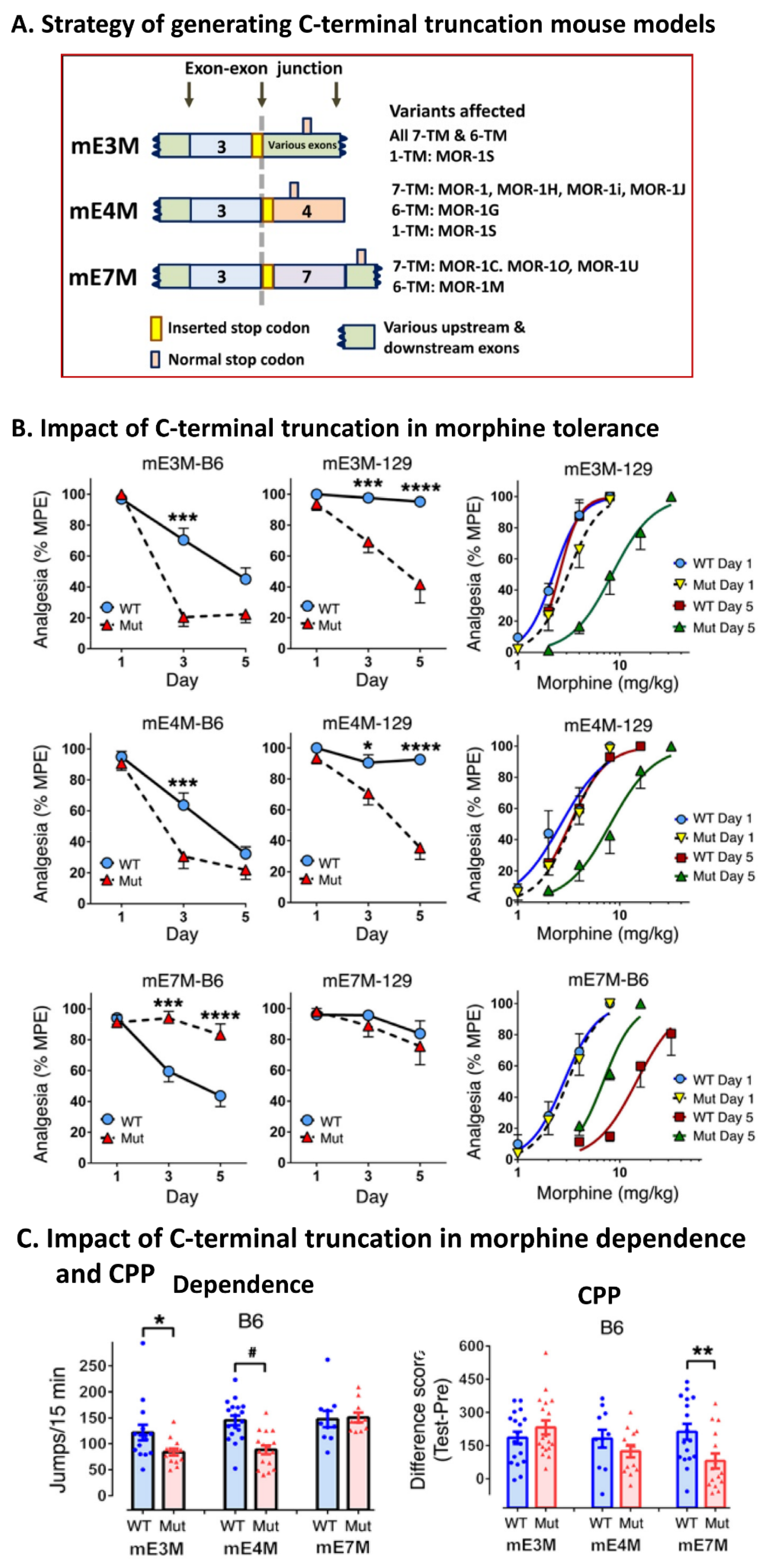
5.2. In Vivo Function of Intracellular C-Terminal Tails: Targeting Alternative Exons Downstream of Exon 3
5.2.1. The Role of Alternative Intracellular C-Terminal Tails in Morphine Tolerance, Physical Dependence, and Reward
5.2.2. The Role of Alternative Intracellular C-Terminal Tails in Morphine Locomotion, Catalepsy, and Inhibition of Gastrointestinal Transit
5.2.3. Involvement of Exon 7-Associated C-Terminal Variants in β-Arrestin 2-Dependent and -Independent Signaling
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mattson, C.L.; Tanz, L.J.; Quinn, K.; Kariisa, M.; Patel, P.; Davis, N.L. Trends and Geographic Patterns in Drug and Synthetic Opioid Overdose Deaths—United States, 2013–2019. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.R. Opioid Antagonists. Pharmacol. Rev. 1967, 19, 463–521. [Google Scholar] [PubMed]
- Pert, C.B.; Pasternak, G.W.; Snyder, S.H. Opiate agonists and antagonists discriminated by receptor binding in brain. Science 1973, 182, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.J.; Hiller, J.M.; Edelman, I. Stereospecific binding of the potent narcotic analgesice [3H]Etorphine to rat-brain homogenate. Proc. Natl. Acad. Sci. USA 1973, 70, 1947–1949. [Google Scholar] [CrossRef] [Green Version]
- Terenius, L. Stereospecific interaction between narcotic analgesics and a synaptic plasma membrane fraction of rat cerebral cortex. Acta Pharmacol. Toxicol. 1973, 32, 317–320. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Childers, S.R.; Snyder, S.H. Opiate analgesia: Evidence for mediation by a subpopulation of opiate receptors. Science 1980, 208, 514–516. [Google Scholar] [CrossRef]
- Wolozin, B.L.; Pasternak, G.W. Classification of multiple morphine and enkephalin binding sites in the central nervous system. Proc. Natl. Acad. Sci. USA 1981, 78, 6181–6185. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.X. Diversity and complexity of the mu opioid receptor gene: Alternative pre-mRNA splicing and promoters. DNA Cell Biol. 2005, 24, 736–750. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Pan, Y.X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, G.W.; Childers, S.R.; Pan, Y.X. Emerging Insights into Mu Opioid Pharmacology. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 258, pp. 89–125. [Google Scholar] [CrossRef]
- Liu, S.; Kang, W.J.; Abrimian, A.; Xu, J.; Cartegni, L.; Majumdar, S.; Hesketh, P.; Bekker, A.; Pan, Y.X. Alternative Pre-mRNA Splicing of the Mu Opioid Receptor Gene, OPRM1: Insight into Complex Mu Opioid Actions. Biomolecules 2021, 11, 1525. [Google Scholar] [CrossRef]
- Pan, Y.-X. Alternative Pre-mRNA Splicing of Mu Opioid Receptor Gene: Molecular Mechanisms Underlying the Complex Actions of Mu Opioids; Belfer, I.D.L., Ed.; Wiley Blackwell: Hoboken, NJ, USA, 2014; pp. 79–98. [Google Scholar]
- Pan, Y.-X.; Pasternak, G.W. Molecular Biology of Mu Opioid Receptors. In Opiate, 2nd ed.; Pasternak, G.W., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 121–160. [Google Scholar]
- Goldstein, A. Opioid peptides (endorphins) in pituitary and brain. Science 1976, 193, 1081–1086. [Google Scholar] [CrossRef]
- Goldstein, A.; Tachibana, S.; Lowney, L.I.; Hunkapiller, M.; Hood, L. Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proc. Natl. Acad. Sci. USA 1979, 76, 6666–6670. [Google Scholar] [CrossRef] [Green Version]
- Birdsall, N.J.M.; Hulme, E.C. C fragment of lipotropin has a high affinity for brain opiate receptors. Nature 1976, 260, 793–795. [Google Scholar]
- Cox, B.M.; Opheim, K.E.; Teschemacher, H.; Goldstein, A. A peptide-like substance from pituitary that acts like morphine. 2. Purification and properties. Life Sci. 1975, 16, 1777–1782. [Google Scholar] [CrossRef]
- Kieffer, B.L.; Befort, K.; Gaveriaux-Ruff, C.; Hirth, C.G. The d-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA 1992, 89, 12048–12052. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.J.; Keith, D.E., Jr.; Morrison, H.; Magendzo, K.; Edwards, R.H. Cloning of a delta opioid receptor by functional expression. Science 1992, 258, 1952–1955. [Google Scholar] [CrossRef]
- Chen, Y.; Mestek, A.; Liu, J.; Hurley, J.A.; Yu, L. Molecular cloning and functional expression of a μ opioid receptor from rat brain. Mol. Pharmacol. 1993, 44, 8–12. [Google Scholar] [CrossRef]
- Thompson, R.C.; Mansour, A.; Akil, H.; Watson, S.J. Cloning and pharmacological characterization of a rat μ opioid receptor. Neuron 1993, 11, 903–913. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.B.; Imai, Y.; Eppler, C.M.; Gregor, P.; Spivak, C.E.; Uhl, G.R. μ opiate receptor: cDNA cloning and expression. Proc. Natl. Acad. Sci. USA 1993, 90, 10230–10234. [Google Scholar] [CrossRef] [Green Version]
- Eppler, C.M.; Hulmes, J.D.; Wang, J.-B.; Johnson, B.; Corbett, M.; Luthin, D.R.; Uhl, G.R.; Linden, J. Purification and partial amino acid sequence of a μ opioid receptor from rat brain. J. Biol. Chem. 1993, 268, 26447–26451. [Google Scholar] [CrossRef]
- Chen, Y.; Mestek, A.; Liu, J.; Yu, L. Molecular cloning of a rat kappa opioid receptor reveals sequence similarities to the μ and d opioid receptors. Biochem. J. 1993, 295, 625–628. [Google Scholar] [CrossRef]
- Li, S.; Zhu, J.; Chen, C.; Chen, Y.-W.; Deriel, J.K.; Ashby, B.; Liu-Chen, L.-Y. Molecular cloning and expression of a rat kappa opioid receptor. Biochem. J. 1993, 295, 629–633. [Google Scholar] [CrossRef]
- Meng, F.; Xie, G.-X.; Thompson, R.C.; Mansour, A.; Goldstein, A.; Watson, S.J.; Akil, H. Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 9954–9958. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.B.; Johnson, P.S.; Persico, A.M.; Hawkins, A.L.; Griffin, C.A.; Uhl, G.R. Human μ opiate receptor: cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Lett. 1994, 338, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Kozak, C.A.; Filie, J.; Adamson, M.C.; Chen, Y.; Yu, L. Murine chromosomal location of the μ and kappa opioid receptor genes. Genomics 1994, 21, 659–661. [Google Scholar] [CrossRef]
- Giros, B.; Pohl, M.; Rochelle, J.M.; Seldin, M.F. Chromosomal localization of opioid peptide and receptor genes in the mouse. Life Sci. 1995, 56, PL369–PL375. [Google Scholar] [CrossRef]
- Kaufman, D.L.; Keith, D.E., Jr.; Anton, B.; Tian, J.; Magendzo, K.; Newman, D.; Tran, T.H.; Lee, D.S.; Wen, C.; Xia, Y.-R.; et al. Characterization of the murine μ opioid receptor gene. J. Biol. Chem. 1995, 270, 15877–15883. [Google Scholar] [CrossRef] [Green Version]
- Cherny, N.; Ripamonti, C.; Pereira, J.; Davis, C.; Fallon, M.; McQuay, H.; Mercadante, S.; Pasternak, G.; Ventafridda, V. Strategies to manage the adverse effects of oral morphine: An evidence-based report. J. Clin. Oncol. 2001, 19, 2542–2554. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, G.W. The pharmacology of mu analgesics: From patients to genes. Neuroscientist 2001, 7, 220–231. [Google Scholar] [CrossRef]
- Inturrisi, C.E. Clinical pharmacology of opioids for pain. Clin. J. Pain 2002, 18, S3–S13. [Google Scholar] [CrossRef]
- Chou, R.; Fanciullo, G.J.; Fine, P.G.; Adler, J.A.; Ballantyne, J.C.; Davies, P.; Donovan, M.I.; Fishbain, D.A.; Foley, K.M.; Fudin, J.; et al. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J. Pain Off. J. Am. Pain Soc. 2009, 10, 113–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternak, G.W. Incomplete cross tolerance and multiple mu opioid peptide receptors. Trends Pharmacol. Sci. 2001, 22, 67–70. [Google Scholar] [CrossRef]
- Kest, B.; Hopkins, E.; Palmese, C.A.; Adler, M.; Mogil, J.S. Genetic variation in morphine analgesic tolerance: A survey of 11 inbred mouse strains. Pharmacol. Biochem. Behav. 2002, 73, 821–828. [Google Scholar] [CrossRef]
- Kest, B.; Palmese, C.A.; Hopkins, E.; Adler, M.; Juni, A.; Mogil, J.S. Naloxone-precipitated withdrawal jumping in 11 inbred mouse strains: Evidence for common genetic mechanisms in acute and chronic morphine physical dependence. Neuroscience 2002, 115, 463–469. [Google Scholar] [CrossRef]
- Klein, G.; Juni, A.; Waxman, A.R.; Arout, C.A.; Inturrisi, C.E.; Kest, B. A survey of acute and chronic heroin dependence in ten inbred mouse strains: Evidence of genetic correlation with morphine dependence. Pharmacol. Biochem. Behav. 2008, 90, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, A.S.; Goodman, R.R. Autoradiographic analysis of mu1, mu2, and delta opioid binding in the central nervous of C57BL/6BY and CXBK (opioid receptor-deficient) mice. Brain Res. 1985, 360, 108–116. [Google Scholar] [CrossRef]
- Goodman, R.R.; Pasternak, G.W. Visualization of mu1 opiate receptors in rat brain using a computerized autoradiographic subtraction technique. Proc. Natl. Acad. Sci. USA 1985, 82, 6667–6671. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, G.W.; Childers, S.R.; Snyder, S.H. Naloxazone, long-acting opiate antagonist: Effects in intact animals and on opiate receptor binding in vitro. J. Pharmacol. Exp. Ther. 1980, 214, 455–462. [Google Scholar]
- Hazum, E.; Chang, K.J.; Cuatrescasas, P.; Pasternak, G.W. Naloxazone irreversibility inhibits the high affinity binding of [124I]D-ala2-D-leu5-enkephalin. Life Sci. 1981, 29, 843–851. [Google Scholar]
- Hahn, E.F.; Carroll-Buatti, M.; Pasternak, G.W. Irreversible opiate agonists and antagonists: The 14-hydroxydihydromorphinone azines. J. Neurosci. 1982, 2, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.C.; Brown, G.P.; Leventhal, L.; Yang, K.; Pasternak, G.W. Novel receptor mechanisms for heroin and morphine-6β-glucuronide analgesia. Neurosci. Lett. 1996, 216, 1–4. [Google Scholar] [CrossRef]
- Brown, G.P.; Yang, K.; Ouerfelli, O.; Standifer, K.M.; Byrd, D.; Pasternak, G.W. 3H-morphine-6β-glucuronide binding in brain membranes and an MOR-1-transfected cell line. J. Pharmacol. Exp. Ther. 1997, 282, 1291–1297. [Google Scholar]
- Brown, G.P.; Yang, K.; King, M.A.; Rossi, G.C.; Leventhal, L.; Chang, A.; Pasternak, G.W. 3-Methoxynaltrexone, a selective heroin/morphine-6β-glucuronide antagonist. FEBS Lett. 1997, 412, 35–38. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.C.; Pan, Y.X.; Brown, G.P.; Pasternak, G.W. Antisense Mapping the Mor-1 Opioid Receptor—Evidence for Alternative Splicing and A Novel Morphine-6-Beta-Glucuronide Receptor. FEBS Lett. 1995, 369, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xu, J.; Xu, M.; Rossi, G.C.; Majumdar, S.; Pasternak, G.W.; Pan, Y.X. Truncated Mu-Opioid Receptors With 6 Transmembrane Domains Are Essential for Opioid Analgesia. Anesth. Analg. 2018, 126, 1050–1057. [Google Scholar] [CrossRef]
- Abrimian, A.; Kraft, T.; Pan, Y.X. Endogenous Opioid Peptides and Alternatively Spliced Mu Opioid Receptor Seven Transmembrane Carboxyl-Terminal Variants. Int. J. Mol. Sci. 2021, 22, 3779. [Google Scholar] [CrossRef]
- Pasternak, D.A.; Pan, L.; Xu, J.; Yu, R.; Xu, M.M.; Pasternak, G.W.; Pan, Y.X. Identification of three new alternatively spliced variants of the rat mu opioid receptor gene: Dissociation of affinity and efficacy. J. Neurochem. 2004, 91, 881–890. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Bolan, E.; Abbadie, C.; Chang, A.; Zuckerman, A.; Rossi, G.; Pasternak, G.W. Identification and characterization of three new alternatively spliced mu-opioid receptor isoforms. Mol. Pharmacol. 1999, 56, 396–403. [Google Scholar] [CrossRef]
- Pan, L.; Xu, J.; Yu, R.; Xu, M.M.; Pan, Y.X.; Pasternak, G.W. Identification and characterization of six new alternatively spliced variants of the human mu opioid receptor gene, Oprm. Neuroscience 2005, 133, 209–220. [Google Scholar] [CrossRef]
- Xu, J.; Xu, M.; Bolan, E.; Gilbert, A.K.; Pasternak, G.W.; Pan, Y.X. Isolating and characterizing three alternatively spliced mu opioid receptor variants: mMOR-1A, mMOR-1O, and mMOR-1P. Synapse 2014, 68, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.X.; Xu, J.; Bolan, E.; Chang, A.; Mahurter, L.; Rossi, G.; Pasternak, G.W. Isolation and expression of a novel alternatively spliced mu opioid receptor isoform, MOR-1F. FEBS Lett. 2000, 466, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.X.; Xu, J.; Bolan, E.; Moskowitz, H.S.; Xu, M.; Pasternak, G.W. Identification of four novel exon 5 splice variants of the mouse mu-opioid receptor gene: Functional consequences of C-terminal splicing. Mol. Pharmacol. 2005, 68, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.J.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into mu-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Koch, T.; Schulz, S.; Pfeiffer, M.; Klutzny, M.; Schroder, H.; Kahl, E.; Hollt, V. C-terminal splice variants of the mouse mu-opioid receptor differ in morphine-induced internalization and receptor resensitization. J. Biol. Chem. 2001, 276, 31408–31414. [Google Scholar] [CrossRef] [Green Version]
- Koch, T.; Schulz, S.; Schroder, H.; Wolf, R.; Raulf, E.; Hollt, V. Carboxyl-terminal splicing of the rat mu opioid receptor modulates agonist-mediated internalization and receptor resensitization. J. Biol. Chem. 1998, 273, 13652–13657. [Google Scholar] [CrossRef] [Green Version]
- Abbadie, C.; Pasternak, G.W. Differential in vivo internalization of MOR-1 and MOR-1C by morphine. NeuroReport 2001, 12, 3069–3072. [Google Scholar] [CrossRef]
- Bolan, E.A.; Pan, Y.X.; Pasternak, G.W. Functional analysis of MOR-1 splice variants of the mouse mu opioid receptor gene Oprm. Synapse 2004, 51, 11–18. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Mahurter, L.; Xu, M.; Gilbert, A.K.; Pasternak, G.W. Identification and characterization of two new human mu opioid receptor splice variants, hMOR-1O and hMOR-1X. Biochem. Biophys. Res. Commun. 2003, 301, 1057–1061. [Google Scholar] [CrossRef]
- Xu, J.; Lu, Z.; Narayan, A.; Le Rouzic, V.P.; Xu, M.; Hunkele, A.; Brown, T.G.; Hoefer, W.F.; Rossi, G.C.; Rice, R.C.; et al. Alternatively spliced mu opioid receptor C termini impact the diverse actions of morphine. J. Clin. Investig. 2017, 127, 1561–1573. [Google Scholar] [CrossRef]
- Narayan, A.; Hunkele, A.; Xu, J.; Bassoni, D.L.; Pasternak, G.W.; Pan, Y.X. Mu Opioids Induce Biased Signaling at the Full-Length Seven Transmembrane C-Terminal Splice Variants of the mu Opioid Receptor Gene, Oprm1. Cell Mol. Neurobiol. 2020, 41, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Tanowitz, M.; Hislop, J.N.; von Zastrow, M. Alternative splicing determines the post-endocytic sorting fate of G-protein-coupled receptors. J. Biol. Chem. 2008, 283, 35614–35621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Xu, J.; Pan, Y.X. A Truncated Six Transmembrane Splice Variant MOR-1G Enhances Expression of the Full-Length Seven Transmembrane mu-Opioid Receptor through Heterodimerization. Mol. Pharmacol. 2020, 98, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, M.; Brown, T.; Rossi, G.C.; Hurd, Y.L.; Inturrisi, C.E.; Pasternak, G.W.; Pan, Y.X. Stabilization of the μ-opioid receptor by truncated single transmembrane splice variants through a chaperone-like action. J. Biol. Chem. 2013, 288, 21211–21227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lu, Z.; Xu, M.; Rossi, G.C.; Kest, B.; Waxman, A.R.; Pasternak, G.W.; Pan, Y.X. Differential expressions of the alternatively spliced variant mRNAs of the micro opioid receptor gene, OPRM1, in brain regions of four inbred mouse strains. PLoS ONE 2014, 9, e111267. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Faskowitz, A.J.; Rossi, G.C.; Xu, M.; Lu, Z.; Pan, Y.X.; Pasternak, G.W. Stabilization of morphine tolerance with long-term dosing: Association with selective upregulation of mu-opioid receptor splice variant mRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbadie, C.; Pan, Y.-X.; Pasternak, G.W. Differential distribution in rat brain of mu opioid receptor carboxy terminal splice variants MOR-1C and MOR-1-like immunoreactivity: Evidence for region-specific processing. J. Comp. Neurol. 2000, 419, 244–256. [Google Scholar] [CrossRef]
- Abbadie, C.; Pan, Y.-X.; Drake, C.T.; Pasternak, G.W. Comparative immunhistochemical distributions of carboxy terminus epitopes from the mu opioid receptor splice variants MOR-1D, MOR-1 and MOR-1C in the mouse and rat central nervous systems. Neuroscience 2000, 100, 141–153. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, Y.X.; Kolesnikov, Y.; Pasternak, G.W. Immunohistochemical labeling of the mu opioid receptor carboxy terminal splice variant mMOR-1B4 in the mouse central nervous system. Brain Res. 2006, 1099, 33–43. [Google Scholar] [CrossRef]
- Dever, S.M.; Xu, R.; Fitting, S.; Knapp, P.E.; Hauser, K.F. Differential expression and HIV-1 regulation of mu-opioid receptor splice variants across human central nervous system cell types. J. Neurovirol. 2012, 18, 181–190. [Google Scholar] [CrossRef]
- Dever, S.M.; Costin, B.N.; Xu, R.; El-Hage, N.; Balinang, J.; Samoshkin, A.; O’Brien, M.A.; McRae, M.; Diatchenko, L.; Knapp, P.E.; et al. Differential expression of the alternatively spliced OPRM1 isoform mu-opioid receptor-1K in HIV-infected individuals. AIDS 2014, 28, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.G.; Xu, J.; Hurd, Y.L.; Pan, Y.X. Dysregulated expression of the alternatively spliced variant mRNAs of the mu opioid receptor gene, OPRM1, in the medial prefrontal cortex of male human heroin abusers and heroin self-administering male rats. J. Neurosci. Res. 2020, 100, 35–47. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, H.; Qin, F.; Wang, Q.; Sun, Q.; Xie, S.; Wang, Q.; Tang, Z.; Lu, Z. Sex Associated Differential Expressions of the Alternatively Spliced Variants mRNA of OPRM1 in Brain Regions of C57BL/6 Mouse. Cell Physiol. Biochem. 2018, 50, 1441–1459. [Google Scholar] [CrossRef]
- Matthes, H.W.D.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; Le Meur, M.; Dollé, P.; et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ opioid-receptor gene. Nature 1996, 383, 819–823. [Google Scholar] [CrossRef]
- Sora, I.; Takahashi, N.; Funada, M.; Ujike, H.; Revay, R.S.; Donovan, D.M.; Miner, L.L.; Uhl, G.R. Opiate receptor knockout mice define μ receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc. Natl. Acad. Sci. USA 1997, 94, 1544–1549. [Google Scholar] [CrossRef] [Green Version]
- Schuller, A.G.; King, M.A.; Zhang, J.; Bolan, E.; Pan, Y.X.; Morgan, D.J.; Chang, A.; Czick, M.E.; Unterwald, E.M.; Pasternak, G.W.; et al. Retention of heroin and morphine-6β-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat. Neurosci. 1999, 2, 151–156. [Google Scholar] [CrossRef]
- Tian, M.; Broxmeyer, H.E.; Fan, Y.; Lai, Z.; Zhang, S.; Aronica, S.; Cooper, S.; Bigsby, R.M.; Steinmetz, R.; Engle, S.J.; et al. Altered hematopoiesis, behavior, and sexual function in μ opioid receptor-deficient mice. J. Exp. Med. 1997, 185, 1517–1522. [Google Scholar] [CrossRef]
- Loh, H.H.; Liu, H.C.; Cavalli, A.; Yang, W.L.; Chen, Y.F.; Wei, L.N. μ opioid receptor knockout in mice: Effects on ligand-induced analgesia and morphine lethality. Mol. Brain Res. 1998, 54, 321–326. [Google Scholar] [CrossRef]
- Erbs, E.; Faget, L.; Scherrer, G.; Matifas, A.; Filliol, D.; Vonesch, J.L.; Koch, M.; Kessler, P.; Hentsch, D.; Birling, M.C.; et al. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct. Funct. 2015, 220, 677–702. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, A.T.; Semache, M.; Gross, F.; Da Fonte, D.F.; Runtz, L.; Colley, C.; Mezni, A.; Le Gouill, C.; Lukasheva, V.; Hogue, M.; et al. Biased Signaling of the Mu Opioid Receptor Revealed in Native Neurons. iScience 2019, 14, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Weibel, R.; Reiss, D.; Karchewski, L.; Gardon, O.; Matifas, A.; Filliol, D.; Becker, J.A.J.; Wood, J.N.; Kieffer, B.L.; Gaveriaux-Ruff, C. Mu Opioid Receptors on Primary Afferent Nav1.8 Neurons Contribute to Opiate-Induced Analgesia: Insight from Conditional Knockout Mice. PLoS ONE 2013, 8, e74706. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.X.; Xu, J.; Xu, M.; Rossi, G.C.; Matulonis, J.E.; Pasternak, G.W. Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. Proc. Natl. Acad. Sci. USA 2009, 106, 4917–4922. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xu, J.; Rossi, G.C.; Majumdar, S.; Pasternak, G.W.; Pan, Y.X. Mediation of opioid analgesia by a truncated 6-transmembrane GPCR. J. Clin. Investig. 2015, 125, 2626–2630. [Google Scholar] [CrossRef] [Green Version]
- Mague, S.D.; Isiegas, C.; Huang, P.; Liu-Chen, L.Y.; Lerman, C.; Blendy, J.A. Mouse model of OPRM1 (A118G) polymorphism has sex-specific effects on drug-mediated behavior. Proc. Natl. Acad. Sci. USA 2009, 106, 10847–10852. [Google Scholar] [CrossRef] [Green Version]
- Ramchandani, V.A.; Umhau, J.; Pavon, F.J.; Ruiz-Velasco, V.; Margas, W.; Sun, H.; Damadzic, R.; Eskay, R.; Schoor, M.; Thorsell, A.; et al. A genetic determinant of the striatal dopamine response to alcohol in men. Mol. Psychiatry 2011, 16, 809–817. [Google Scholar] [CrossRef]
- Wang, X.F.; Barbier, E.; Chiu, Y.T.; He, Y.; Zhan, J.; Bi, G.H.; Zhang, H.Y.; Feng, B.; Liu-Chen, L.Y.; Wang, J.B.; et al. T394A Mutation at the mu Opioid Receptor Blocks Opioid Tolerance and Increases Vulnerability to Heroin Self-Administration in Mice. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 10392–10403. [Google Scholar] [CrossRef] [Green Version]
- Grecksch, G.; Just, S.; Pierstorff, C.; Imhof, A.K.; Gluck, L.; Doll, C.; Lupp, A.; Becker, A.; Koch, T.; Stumm, R.; et al. Analgesic tolerance to high-efficacy agonists but not to morphine is diminished in phosphorylation-deficient S375A mu-opioid receptor knock-in mice. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 13890–13896. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 2019, 10, 367. [Google Scholar] [CrossRef]
- Charbogne, P.; Gardon, O.; Martin-Garcia, E.; Keyworth, H.L.; Matsui, A.; Mechling, A.E.; Bienert, T.; Nasseef, T.; Robe, A.; Moquin, L.; et al. Mu Opioid Receptors in Gamma-Aminobutyric Acidergic Forebrain Neurons Moderate Motivation for Heroin and Palatable Food. Biol. Psychiatry 2017, 81, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Boulos, L.J.; Ben Hamida, S.; Bailly, J.; Maitra, M.; Ehrlich, A.T.; Gaveriaux-Ruff, C.; Darcq, E.; Kieffer, B.L. Mu opioid receptors in the medial habenula contribute to naloxone aversion. Neuropsychopharmacology 2020, 45, 247–255. [Google Scholar] [CrossRef]
- Sun, J.; Chen, S.R.; Pan, H.L. mu-Opioid receptors in primary sensory neurons are involved in supraspinal opioid analgesia. Brain Res. 2020, 1729, 146623. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.L.; Perez, C.T.; Halemani, D.N.; Shao, X.M.; Greenwood, M.; Heath, S.; Feldman, J.L.; Kam, K. Opioids modulate an emergent rhythmogenic process to depress breathing. eLife 2019, 8, e50613. [Google Scholar] [CrossRef] [PubMed]
- Severino, A.L.; Mittal, N.; Hakimian, J.K.; Velarde, N.; Minasyan, A.; Albert, R.; Torres, C.; Romaneschi, N.; Johnston, C.; Tiwari, S.; et al. mu-Opioid Receptors on Distinct Neuronal Populations Mediate Different Aspects of Opioid Reward-Related Behaviors. eNeuro 2020, 7, 0146. [Google Scholar] [CrossRef] [PubMed]
- Inturrisi, C.E.; Yoburn, B.C.; Portenoy, R.K.; Foley, K.M. Species dependent formation of morphine-6-glucuronide (M-6-G) from morphine (MOR). Comm. Probl. Drug Depend. 1996, 174, 157. [Google Scholar]
- Pan, Y.X.; Xu, A.; Mahurter, L.; Bolan, E.; Xu, M.M.; Pasternak, G.W. Generation of the mu opioid receptor (MOR-1) protein by three new splice variants of the Oprm gene. Proc. Natl. Acad. Sci. USA 2001, 98, 14084–14089. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.C.; Leventhal, L.; Pan, Y.X.; Cole, J.; Su, W.; Bodnar, R.J.; Pasternak, G.W. Antisense mapping of MOR-1 in the rat: Distinguishing between morphine and morphine-6β-glucuronide antinociception. J. Pharmacol. Exp. Ther. 1997, 281, 109–114. [Google Scholar]
- Kitanaka, N.; Sora, I.; Kinsey, S.; Zeng, Z.; Uhl, G.R. No heroin or morphine 6β-glucuronide analgesia in mu-opioid receptor knockout mice. Eur. J. Pharmacol. 1998, 355, R1–R3. [Google Scholar] [CrossRef]
- Majumdar, S.; Grinnell, S.; Le, R.V.; Burgman, M.; Polikar, L.; Ansonoff, M.; Pintar, J.; Pan, Y.X.; Pasternak, G.W. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc. Natl. Acad. Sci. USA 2011, 108, 19778–19783. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, S.; Subrath, J.; Le, R.V.; Polikar, L.; Burgman, M.; Nagakura, K.; Ocampo, J.; Haselton, N.; Pasternak, A.R.; Grinnell, S.; et al. Synthesis and Evaluation of Aryl-Naloxamide Opiate Analgesics Targeting Truncated Exon 11-Associated mu Opioid Receptor (MOR-1) Splice Variants. J. Med. Chem. 2012, 55, 6352–6362. [Google Scholar] [CrossRef] [Green Version]
- Wieskopf, J.S.; Pan, Y.X.; Marcovitz, J.; Tuttle, A.H.; Majumdar, S.; Pidakala, J.; Pasternak, G.W.; Mogil, J.S. Broad-spectrum analgesic efficacy of IBNtxA is mediated by exon 11-associated splice variants of the mu-opioid receptor gene. Pain 2014, 155, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- Marrone, G.F.; Grinnell, S.G.; Lu, Z.; Rossi, G.C.; Le Rouzic, V.; Xu, J.; Majumdar, S.; Pan, Y.X.; Pasternak, G.W. Truncated mu opioid GPCR variant involvement in opioid-dependent and opioid-independent pain modulatory systems within the CNS. Proc. Natl. Acad. Sci. USA 2016, 113, 3663–3668. [Google Scholar] [CrossRef] [Green Version]
- Grinnell, S.G.; Ansonoff, M.; Marrone, G.F.; Lu, Z.; Narayan, A.; Xu, J.; Rossi, G.; Majumdar, S.; Pan, Y.X.; Bassoni, D.L.; et al. Mediation of buprenorphine analgesia by a combination of traditional and truncated mu opioid receptor splice variants. Synapse 2016, 70, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Marrone, G.F.; Lu, Z.; Rossi, G.; Narayan, A.; Hunkele, A.; Marx, S.; Xu, J.; Pintar, J.; Majumdar, S.; Pan, Y.X.; et al. Tetrapeptide Endomorphin Analogs Require Both Full Length and Truncated Splice Variants of the Mu Opioid Receptor Gene Oprm1 for Analgesia. ACS Chem. Neurosci. 2016, 7, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Gomes, I.; Ayoub, M.A.; Fujita, W.; Jaeger, W.C.; Pfleger, K.D.; Devi, L.A. G Protein-Coupled Receptor Heteromers. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 403–425. [Google Scholar] [CrossRef] [Green Version]
- Samoshkin, A.; Convertino, M.; Viet, C.T.; Wieskopf, J.S.; Kambur, O.; Marcovitz, J.; Patel, P.; Stone, L.S.; Kalso, E.; Mogil, J.S.; et al. Structural and functional interactions between six-transmembrane mu-opioid receptors and β2-adrenoreceptors modulate opioid signaling. Sci. Rep. 2015, 5, 18198. [Google Scholar] [CrossRef]
- Lejeune, F.; Maquat, L.E. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr. Opin. Cell Biol. 2005, 17, 309–315. [Google Scholar] [CrossRef]
- Chang, Y.F.; Imam, J.S.; Wilkinson, M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef] [Green Version]
- Tanowitz, M.; von Zastrow, M. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J. Biol. Chem. 2003, 278, 45978–45986. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170, e413. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.L.R.V.P.; Quadros, R.M.; Pasternak, G.W.; Yoshiki, M.; Mashimo, T.; Saunders, T.L.; Gurumurthy, C.B.; Pan, Y.-X. Generation of conditional Oprm1 knockout rat models using Easi-CRISPR with long ssDNA donors. In Proceedings of the International Narcotics Research Conference, San Diego, CA, USA, 13–15 June 2018; p. 74. [Google Scholar]
- Raehal, K.M.; Bohn, L.M. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 2011, 60, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Sotnikova, T.D.; Medvedev, I.O.; Lefkowitz, R.J.; Dykstra, L.A.; Caron, M.G. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J. Neurosci. 2003, 23, 10265–10273. [Google Scholar] [CrossRef] [Green Version]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 10494–10500. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Name_Vendor_Stock # | Targeting Site | ES Cell_Strain | Recipient | Lab | Reference |
|---|---|---|---|---|---|---|
| MOR-KO | Oprm1tm1Kff_Jackson Lab_007559 | A neo cassette was inserted in exon 2. | D3/129S2/SvPas | C57BL/6 | Kieffer | [77] |
| MOR-mCherry | Oprm1tm4Kff_Jackson Lab_029013 | mCherry was fused at the end of exon 4. | 129svPas-derived | C57BL/6J | Kieffer | [82] |
| MOR-Venus | Oprm1tm1.1lcs/KffJ_Jackson Lab_035787 | Venus was fused at the end of exon 4. | 129svPas-derived | C57BL/6NCrl | Kieffer | [83] |
| Oprm1Cre:GFP | Oprm1tm1.1(cre/GFP)Rpa_Jackson Lab_035574 | A Cre:GFP/SV40pA cassette was inserted at 5′ of the exon 1 ATG start-codon. | 129S6/SvEvTac-derived | C57BL/6NCrl | Kieffer | N/A |
| Oprm1fl/fl (conditional KO) | Oprm1tm1.1Cgrf/KffJ_Jackson Lab_030074 | Exons 2/3 were floxed with loxPs. | 129Sv-derived | C57BL/6J | Kieffer | [84] |
| Exon-1 MOR-1 KO | Oprm1tm1Uhl_MGI_363132 | A 3.2 kb BglII-EcoRI exon 1-containing region was replaced by a neo cassette. | AB1/129S7/SvEvBrd-Hprt | C57BL/6J | Uhl | [78] |
| Exon-1 MOR-1 KO | N/A | A 2.3 kb BamHI-HindIII exon 1-containing region was replaced by a neo cassette. | CCE/129S/SvEv | C57BL/6 | Pintar | [79] |
| Exon-1 MOR-1 KO | N/A | Exon 1 was replaced by a neo cassette. | D3/129S2/SvPas | C57BL/6J | Yu | [80] |
| Exons 2/3 MOR-1 KO | N/A | Exons 2/3 were replaced by a neo cassette. | HM1/129P2/OlaHsd | C57BL/6J | Loh | [81] |
| Exon 11 KO | N/A | Exon 11 was replaced by a LacZ/neo cassette. | E14/129P2/OlaHsd | C57BL/6J | Pan | [85] |
| Exons 1/11 double KO | N/A | Exon 1 and exon 11 were replaced by ZsGreen/SVpA and tdTomamto/BGHpA, respectively. | W4/129S6/SvEvTac | C57BL/6J | Pan | [86] |
| mE3M-129 | 129S6(B6)-Oprm1tm3.1Yxp/Mmmh_ MMRRC_65820 | A stop codon was inserted at the end of exon 3 in 129S6/SvEvTac. | W4/129S6/SvEvTac | 129S6/SvEvTac | Pan | [63] |
| mE3M-B6 | B6(Cg)-Oprm1tm3.1Yxp/Mmmh_ MMRRC_625821 | A stop codon was inserted at the end of exon 3 in C57BL/6J. | CY2.4/B6(Cg)-Tyr | C57BL/6J | Pan | [63] |
| mE4M-129 | 129S6(B6)-Oprm1tm4.1Yxp/Mmmh_ MMRRC_65823 | A stop codon was inserted at the beginning of exon 4 in 129S6/Sv/EvTac. | W4/129S6/SvEvTac | 129S6/SvEvTac | Pan | [63] |
| mE4M-B6 | B6J.129S6-Oprm1tm4.1Yxp/Mmmh_ MMRRC_65822 | A stop codon was inserted at the beginning of exon 4 in C57BL/6J. | W4/129S6/SvEvTac | C57BL/6J | Pan | [63] |
| mE7M-129 | B6J.129S6-Oprm1tm2.1Yxp/Mmmh_ MMRRC_65824 | A stop codon was inserted at the beginning of exon 7 in C57BL/6J. | W4/129S6/SvEvTac | 129S6/SvEvTac | Pan | [63] |
| mE7M-B6 | 129S6(B6)-Oprm1tm2.1Yxp/Mmmh_ MMRRC_65833 | A stop codon was inserted at the beginning of exon 7 in C57BL/6J. | W4/129S6/SvEvTac | C57BL/6J | Pan | [63] |
| MOR-KI (A112G) | Oprm1tm1jabl_MGI_3810212 | Point mutation of mouse Oprm1 SNP (A112G) that mimicked the human SNP A118G | ES cell/C57BL/6 (Chemicon) | C57BL/6 | Blendy | [87] |
| Humanized OPRM1-118A /OPRM1-118G | C57BL/6-Oprm1tm1.1Arte: 118AC57BL_6-Oprm1tm2.1Arte: 118G | Part of mouse exon 1 was replaced by corresponding human exon 1 with the SNP. | ES cell/C57BL/6NTac | C57BL/6 | Heilig | [88] |
| MOR-KI (T394A) | Oprm1tm1.1jbwa_Jackson Lab_026221 | Point mutation at T394 | ES cell/129S6/SvEvTac (inGenious Targeting Laboratory) | C57BL/6NTac | Wang | [89] |
| MOP KI (S375A) | Oprm1tm1Shlz_MGI_5000465 | Point mutation at S375 | Bruce 4/B6.Cg-Thy1 | C57BL/6J | Schulz | [90] |
| MOP KI (10S/T-A) | Oprm1tm2.1Shlz_MGI_6117668 | 10 serine/threonine phosphorylation sites within the 354–394 region were mutated to alanine. | Bruce 4/B6.Cg-Thy1 | C57BL/6J | Schulz | [91] |
| MOP KI (11S/T-A) | Oprm1tm3.Shlz_MGI_6117673 | 11 serine/threonine phosphorylation sites within the 354–383 region were mutated to alanine. | Bruce 4/B6.Cg-Thy1 | C57BL/6J | Schulz | [91] |
| Classification | Drug | Analgesia | References | ||||
|---|---|---|---|---|---|---|---|
| E1-KO | E11-KO | E1/E11-KO | E1/E11-KO + Lenti-6TM | E11-KO + Lenti-6TM | |||
| 7TM variant- dependent | Morphine | Lost | Retained | Lost | N/A | N/A | [79,85] |
| Methadone | Lost | Retained | Lost | N/A | N/A | [79,85] | |
| DAMGO | Lost | Retained | N/A | N/A | N/A | [106] | |
| 7TM + 6TM variant- dependent | Buprenorphine | Lost | Lost | Lost | Not rescued | Rescued | [86,105] |
| M6G | Retained but reduced | Reduced | N/A | N/A | N/A | [79,85] | |
| Heroin | Retained but reduced | Reduced | N/A | N/A | N/A | [79,85] | |
| DAPP | N/A | Lost | N/A | Not rescued | Rescued | [106] | |
| IDAPP | N/A | Lost | N/A | Not rescued | Rescued | [106] | |
| DPDPE | N/A | Lost | N/A | Not rescued | Rescued | [107] | |
| SNC80 | N/A | Lost | N/A | Not rescued | Rescued | [107] | |
| 6TM variant- dependent | IBNtxA | N/A | Lost | Lost | Rescued | N/A | [86,101] |
| Ketocyclazocin | N/A | Lost | Lost | Rescued | N/A | [86,101] | |
| U50,488H | N/A | Lost | Lost | Rescued | Rescued | [107] | |
| Clonidine | N/A | Lost | Lost | Rescued | Rescued | [107] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, W.; Liu, S.; Xu, J.; Abrimian, A.; Malik, A.F.; Chien, R.; Adaralegbe, A.; Amponsah, A.; Cartegni, L.; Pintar, J.; et al. Exploring Pharmacological Functions of Alternatively Spliced Variants of the Mu Opioid Receptor Gene, Oprm1, via Gene-Targeted Animal Models. Int. J. Mol. Sci. 2022, 23, 3010. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063010
Kang W, Liu S, Xu J, Abrimian A, Malik AF, Chien R, Adaralegbe A, Amponsah A, Cartegni L, Pintar J, et al. Exploring Pharmacological Functions of Alternatively Spliced Variants of the Mu Opioid Receptor Gene, Oprm1, via Gene-Targeted Animal Models. International Journal of Molecular Sciences. 2022; 23(6):3010. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063010
Chicago/Turabian StyleKang, Wenjian, Shan Liu, Jin Xu, Anna Abrimian, Ayma F. Malik, Raymond Chien, Adejuyigbe Adaralegbe, Akwasi Amponsah, Luca Cartegni, John Pintar, and et al. 2022. "Exploring Pharmacological Functions of Alternatively Spliced Variants of the Mu Opioid Receptor Gene, Oprm1, via Gene-Targeted Animal Models" International Journal of Molecular Sciences 23, no. 6: 3010. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063010