
Host Restriction Factors Modulating HIV Latency and Replication in Macrophages

 and
and
Abstract
:1. Introduction on Macrophages as Targets of HIV Replication
2. Host Cell Restriction Factors, HIV Infection and Replication
3. Viral Proteins Counteracting Restriction Factors
4. Pharmacological Targeting of HIV Proteins
5. Macrophage Polarization to a Pro-Inflammatory Mode: A Model of HIV-1 Restriction
6. Exploiting M1 Polarization to Generate a Model of Reversible HIV-1 Latency in Primary MDM
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Pablo-Maiso, L.; Doménech, A.; Echeverría, I.; Gómez-Arrebola, C.; De Andrés, D.; Rosati, S.; Gómez-Lucia, E.; Reina, R. Prospects in Innate Immune Responses as Potential Control Strategies against Non-Primate Lentiviruses. Viruses 2018, 10, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.W.; Churchill, M.; Di Mascio, M.; Katlama, C.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [PubMed]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2015, 17, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeffel, G.; Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol. 2018, 330, 5–15. [Google Scholar] [CrossRef]
- Yip, J.L.; Balasuriya, G.K.; Spencer, S.J.; Hill-Yardin, E.L. The Role of Intestinal Macrophages in Gastrointestinal Homeostasis: Heterogeneity and Implications in Disease. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1701–1718. [Google Scholar] [CrossRef]
- Shi, T.; Denney, L.; An, H.; Ho, L.; Zheng, Y. Alveolar and lung interstitial macrophages: Definitions, functions, and roles in lung fibrosis. J. Leukoc. Biol. 2020, 110, 107–114. [Google Scholar] [CrossRef]
- Brew, B.J.; Barnes, S.L. The impact of HIV central nervous system persistence on pathogenesis. AIDS 2019, 33, S113–S121. [Google Scholar] [CrossRef]
- Hendricks, C.M.; Cordeiro, T.; Gomes, A.P.; Stevenson, M. The Interplay of HIV-1 and Macrophages in Viral Persistence. Front. Microbiol. 2021, 12, 646447. [Google Scholar] [CrossRef]
- Beck, S.E.; Queen, S.E.; Pate, K.A.M.; Mangus, L.M.; Abreu, C.M.; Gama, L.; Witwer, K.; Adams, R.J.; Zink, M.C.; Clements, J.E.; et al. An SIV/macaque model targeted to study HIV-associated neurocognitive disorders. J. Neurovirol. 2017, 24, 204–212. [Google Scholar] [CrossRef]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Summers, K.M.; Rehli, M. Transcriptional Regulation and Macrophage Differentiation. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, I.A.T.M.; Porterfield, J.Z.; Gupta, R.K.; Mlcochova, P. Cell Cycle Regulation in Macrophages and Susceptibility to HIV-1. Viruses 2020, 12, 839. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, D.; Li, G. The role of microglia in viral encephalitis: A review. J. Neuroinflamm. 2019, 16, 76. [Google Scholar] [CrossRef]
- Tan, J.; Sattentau, Q.J. The HIV-1-containing macrophage compartment: A perfect cellular niche? Trends Microbiol. 2013, 21, 405–412. [Google Scholar] [CrossRef]
- Graziano, F.; Vicenzi, E.; Poli, G. The ATP/P2X7 axis in human immunodeficiency virus infection of macrophages. Curr. Opin. Pharmacol. 2019, 47, 46–52. [Google Scholar] [CrossRef]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.-A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef]
- Costiniuk, C.T.; Jenabian, M.-A. The lungs as anatomical reservoirs of HIV infection. Rev. Med. Virol. 2013, 24, 35–54. [Google Scholar] [CrossRef]
- Lamers, S.L.; Rose, R.; Maidji, E.; Agsalda-Garcia, M.; Nolan, D.J.; Fogel, G.B.; Salemi, M.; Garcia, D.L.; Bracci, P.; Yong, W.; et al. HIV DNA Is Frequently Present within Pathologic Tissues Evaluated at Autopsy from Combined Antiretroviral Therapy-Treated Patients with Undetectable Viral Loads. J. Virol. 2016, 90, 8968–8983. [Google Scholar] [CrossRef] [Green Version]
- Veenhuis, R.T.; Abreu, C.M.; Shirk, E.N.; Gama, L.; Clements, J.E. HIV replication and latency in monocytes and macrophages. Semin. Immunol. 2021, 51, 101472. [Google Scholar] [CrossRef]
- Chintala, K.; Mohareer, K.; Banerjee, S. Dodging the Host Interferon-Stimulated Gene Mediated Innate Immunity by HIV-1: A Brief Update on Intrinsic Mechanisms and Counter-Mechanisms. Front. Immunol. 2021, 12, 716927. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.; Choi, J.D.; Malim, M. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Deeks, S.G.; Archin, N.; Cannon, P.; Collins, S.; Jones, R.B.; de Jong, M.A.W.P.; Lambotte, O.; Lamplough, R.; Ndung’U, T.; Sugarman, J.; et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nat. Med. 2021, 27, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM Proteins Incorporated into HIV-1 Virions Impair Viral Fusion and Spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-Inducible Cholesterol-25-Hydroxylase Broadly Inhibits Viral Entry by Production of 25-Hydroxycholesterol. Immunity 2012, 38, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, M.; Hsieh, W.Y.; Robertson, K.A.; Kropp, K.A.; Forster, T.; Shui, G.; Lacaze, P.; Watterson, S.; Griffiths, S.J.; Spann, N.J.; et al. The Transcription Factor STAT-1 Couples Macrophage Synthesis of 25-Hydroxycholesterol to the Interferon Antiviral Response. Immunity 2013, 38, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabryova, H.; Strebel, K. Vpr and Its Cellular Interaction Partners: R We There Yet? Cells 2019, 8, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotard, M.; Tsopoulidis, N.; Tibroni, N.; Willemsen, J.; Binder, M.; Ruggieri, A.; Fackler, O.T. Sensing of HIV-1 Infection in Tzm-bl Cells with Reconstituted Expression of STING. J. Virol. 2016, 90, 2064–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Rui, Y.; Lou, M.; Yin, L.; Xiong, H.; Zhou, Z.; Shen, S.; Chen, T.; Zhang, Z.; Zhao, N. HIV-2/SIV Vpx targets a novel functional domain of STING to selectively inhibit cGAS-STING-mediated NF-kappaB signalling. Nat. Microbiol. 2019, 4, 2552–2564. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.M.; Marno, K.M.; Pike, R.; Lee, W.-Y.; Jones, C.E.; Ogunkolade, B.W.; Pardieu, C.; Bryan, A.; Fu, R.M.; Warnes, G.; et al. HIV-1 Accessory Protein Vpr Interacts with REAF/RPRD2 To Mitigate Its Antiviral Activity. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Cloherty, A.; Rader, A.; Compeer, B.; Ribeiro, C. Human TRIM5α: Autophagy Connects Cell-Intrinsic HIV-1 Restriction and Innate Immune Sensor Functioning. Viruses 2021, 13, 320. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Dauphin, A.; Komurlu, S.; McCauley, S.M.; Yurkovetskiy, L.; Carbone, C.; Diehl, W.E.; De Castillia, C.S.; Campbell, E.M.; Luban, J. Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat. Microbiol. 2019, 4, 2044–2051. [Google Scholar] [CrossRef]
- Selyutina, A.; Persaud, M.; Simons, L.M.; Bulnes-Ramos, A.; Buffone, C.; Martinez-Lopez, A.; Scoca, V.; Di Nunzio, F.; Hiatt, J.; Marson, A.; et al. Cyclophilin A Prevents HIV-1 Restriction in Lymphocytes by Blocking Human TRIM5α Binding to the Viral Core. Cell Rep. 2020, 30, 3766–3777. [Google Scholar] [CrossRef]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Su, L. Vpr Enhances HIV-1 Env Processing and Virion Infectivity in Macrophages by Modulating TET2-Dependent IFITM3 Expression. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Wheeler, L.A.; Trifonova, R.T.; Vrbanac, V.; Barteneva, N.S.; Liu, X.; Bollman, B.; Onofrey, L.; Mulik, S.; Ranjbar, S.; Luster, A.D.; et al. TREX1 Knockdown Induces an Interferon Response to HIV that Delays Viral Infection in Humanized Mice. Cell Rep. 2016, 15, 1715–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, N.; Regalado-Magdos, A.D.; Stiggelbout, B.; Lee-Kirsch, M.A.; Lieberman, J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010, 11, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Jin, T.; Laustsen, A.; Hansen, K.; Østergaard, L.; Fitzgerald, K.A.; et al. PNAS Plus: From the Cover: IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, K.; Wang, Z.; Pan, Q.; Tan, J.; Qiao, W.; Liang, C. Effect of Different Nuclear Localization Signals on the Subcellular Localization and Anti-HIV-1 Function of the MxB Protein. Front. Microbiol. 2021, 12, 1241. [Google Scholar] [CrossRef]
- Pagani, I.; Poli, G.; Vicenzi, E. TRIM22. A Multitasking Antiviral Factor. Cells 2021, 10, 1864. [Google Scholar] [CrossRef]
- Kajaste-Rudnitski, A.; Marelli, S.S.; Pultrone, C.; Pertel, T.; Uchil, P.D.; Mechti, N.; Mothes, W.; Poli, G.; Luban, J.; Vicenzi, E. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-κB-responsive long terminal repeat elements. J. Virol. 2011, 85, 5183–5196. [Google Scholar] [CrossRef] [Green Version]
- Montano, M.A.; Kripke, K.; Norina, C.D.; Achacoso, P.; Herzenberg, L.A.; Roy, A.L.; Nolan, G.P. NF-kappa B homodimer binding within the HIV-1 initiator region and interactions with TFII-I. Proc. Natl. Acad. Sci. USA 1996, 93, 12376–12381. [Google Scholar] [CrossRef] [Green Version]
- Della Chiara, G.; Crotti, A.; Liboi, E.; Giacca, M.; Poli, G.; Lusic, M. Negative regulation of HIV-1 transcription by a heterodimeric NF-kappaB1/p50 and C-terminally truncated STAT5 complex. J. Mol. Biol. 2011, 410, 933–943. [Google Scholar] [CrossRef]
- Allouch, A.; David, A.; Amie, S.M.; Lahouassa, H.; Chartier, L.; Margottin-Goguet, F.; Barré-Sinoussi, F.; Kim, B.; Sáez-Cirión, A.; Pancino, G. p21-mediated RNR2 repression restricts HIV-1 replication in macrophages by inhibiting dNTP biosynthesis pathway. Proc. Natl. Acad. Sci. USA 2013, 110, E3997–E4006. [Google Scholar] [CrossRef] [Green Version]
- Valle-Casuso, J.C.; Allouch, A.; David, A.; Lenzi, G.M.; Studdard, L.; Barré-Sinoussi, F.; Müller-Trutwin, M.; Kim, B.; Pancino, G.; Sáez-Cirión, A. p21 Restricts HIV-1 in Monocyte-Derived Dendritic Cells through the Reduction of Deoxynucleoside Triphosphate Biosynthesis and Regulation of SAMHD1 Antiviral Activity. J. Virol. 2017, 91, e01324-17. [Google Scholar] [CrossRef] [Green Version]
- Graziano, F.; Aimola, G.; Forlani, G.; Turrini, F.; Accolla, R.S.; Vicenzi, E.; Poli, G. Reversible Human Immunodeficiency Virus Type-1 Latency in Primary Human Monocyte-Derived Macrophages Induced by Sustained M1 Polarization. Sci. Rep. 2018, 8, 14249. [Google Scholar] [CrossRef] [PubMed]
- Forlani, G.; Shallak, M.; Ramia, E.; Tedeschi, A.; Accolla, R.S. Restriction factors in human retrovirus infections and the unprecedented case of CIITA as link of intrinsic and adaptive immunity against HTLV-1. Retrovirology 2019, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Chougui, G.; Munir-Matloob, S.; Matkovic, R.; Martin, M.M.; Morel, M.; Lahouassa, H.; LeDuc, M.; Ramirez, B.C.; Etienne, L.; Margottin-Goguet, F. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat. Microbiol. 2018, 3, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Nodder, S.B.; Gummuluru, S. Illuminating the Role of Vpr in HIV Infection of Myeloid Cells. Front. Immunol. 2019, 10, 1606. [Google Scholar] [CrossRef]
- Chu, H.; Wang, J.-J.; Qi, M.; Yoon, J.-J.; Chen, X.; Wen, X.; Hammonds, J.; Ding, L.; Spearman, P. Tetherin/BST-2 Is Essential for the Formation of the Intracellular Virus-Containing Compartment in HIV-Infected Macrophages. Cell Host Microbe 2012, 12, 360–372. [Google Scholar] [CrossRef] [Green Version]
- Giese, S.; Lawrence, S.P.; Mazzon, M.; Nijmeijer, B.M.; Marsh, M. The Nef Protein of the Macrophage Tropic HIV-1 Strain AD8 Counteracts Human BST-2/Tetherin. Viruses 2020, 12, 459. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The Interferon-Induced Protein BST-2 Restricts HIV-1 Release and Is Downregulated from the Cell Surface by the Viral Vpu Protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Galao, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J. Innate sensing of HIV-1 assembly by Tetherin induces NFkappaB-dependent proinflammatory responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Krapp, C.; Hotter, D.; Gawanbacht, A.; McLaren, P.J.; Kluge, S.F.; Stürzel, C.M.; Mack, K.; Reith, E.; Engelhart, S.; Ciuffi, A.; et al. Guanylate Binding Protein (GBP) 5 Is an Interferon-Inducible Inhibitor of HIV-1 Infectivity. Cell Host Microbe 2016, 19, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, T.; Zhang, Y.; Fujita, H.; Tokunaga, K. MARCH8: The tie that binds to viruses. FEBS J. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Zhang, Y.; Koyama, T.; Tobiume, M.; Tsunetsugu-Yokota, Y.; Yamaoka, S.; Fujita, H.; Tokunaga, K. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat. Med. 2015, 21, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tada, T.; Ozono, S.; Yao, W.; Tanaka, M.; Yamaoka, S.; Kishigami, S.; Fujita, H.; Tokunaga, K. Membrane-associated RING-CH (MARCH) 1 and 2 are MARCH family members that inhibit HIV-1 infection. J. Biol. Chem. 2019, 294, 3397–3405. [Google Scholar] [CrossRef] [Green Version]
- Lubow, J.; Virgilio, M.C.; Merlino, M.; Collins, D.R.; Mashiba, M.; Peterson, B.G.; Lukic, Z.; Painter, M.M.; Gomez-Rivera, F.; Terry, V.; et al. Mannose receptor is an HIV restriction factor counteracted by Vpr in macrophages. eLife 2020, 9, e51035. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Egan, B.S.; Shepherd, V.L. HIV-1 Nef mediates post-translational down-regulation and redistribution of the mannose receptor. J. Leukoc. Biol. 2005, 77, 522–534. [Google Scholar] [CrossRef]
- Balliet, J.W.; Kolson, D.L.; Eiger, G.; Kim, F.M.; McGann, K.A.; Srinivasan, A.; Collman, R. Distinct Effects in Primary Macrophages and Lymphocytes of the Human Immunodeficiency Virus Type 1 Accessory Genes vpr, vpu, and nef: Mutational Analysis of a Primary HIV-1 Isolate. Virology 1994, 200, 623–631. [Google Scholar] [CrossRef]
- Mashiba, M.; Collins, D.R.; Terry, V.H.; Collins, K.L. Vpr overcomes macrophage-specific restriction of HIV-1 Env expression and virion production. Cell Host Microbe 2014, 16, 722–735. [Google Scholar] [CrossRef] [Green Version]
- Muthumani, K.; Choo, A.Y.; Zong, W.X.; Madesh, M.; Hwang, D.S.; Premkumar, A.; Thieu, K.P.; Emmanuel, J.; Kumar, S.; Thompson, C.B.; et al. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nat. Cell Biol. 2006, 8, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Jowett, J.B.; Planelles, V.; Poon, B.; Shah, N.P.; Chen, M.L.; Chen, I.S. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 1995, 69, 6304–6313. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, M.A.; Koepp, D.M.; Silver, P.A.; Emerman, M. HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev. 1998, 12, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.M.; Weeger, M.; Stahl-Hennig, C.; Coulibaly, C.; Hunsmann, G.; Muller, J.; Muller-Hermelink, H.; Fuchs, D.; Wachter, H.; Daniel, M.M.; et al. Importance of vpr for infection of rhesus monkeys with simian immunodeficiency virus. J. Virol. 1993, 67, 902–912. [Google Scholar] [CrossRef] [Green Version]
- Hadi, K.; Walker, L.A.; Guha, D.; Murali, R.; Watkins, S.; Tarwater, P.; Srinivasan, A.; Ayyavoo, V. Human immunodeficiency virus type 1 Vpr polymorphisms associated with progressor and nonprogressor individuals alter Vpr-associated functions. J. Gen. Virol. 2014, 95, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaumont, T.; van Nuenen, A.; Broersen, S.; Blattner, W.A.; Lukashov, V.V.; Schuitemaker, H. Reversal of Human Immunodeficiency Virus Type 1 IIIB to a Neutralization-Resistant Phenotype in an Accidentally Infected Laboratory Worker with a Progressive Clinical Course. J. Virol. 2001, 75, 2246–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Wang, Y.; Tokunaga, K.; Huang, F.; Sun, B.; Yang, R. The HIV-1 accessory protein Vpr induces the degradation of the anti-HIV-1 agent APOBEC3G through a VprBP-mediated proteasomal pathway. Virus Res. 2015, 195, 25–34. [Google Scholar] [CrossRef]
- An, J.; Rao, A.; Ko, M. TET family dioxygenases and DNA demethylation in stem cells and cancers. Exp. Mol. Med. 2017, 49, e323. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Wang, Q.; Xu, Y.; Tsao, L.-C.; Nakagawa, T.; Guo, H.; Su, L.; Xiong, Y. Vpr Targets TET2 for Degradation by CRL4VprBP E3 Ligase to Sustain IL-6 Expression and Enhance HIV-1 Replication. Mol. Cell 2018, 70, 961–970.e5. [Google Scholar] [CrossRef] [Green Version]
- Doehle, B.P.; Hladik, F.; McNevin, J.P.; McElrath, M.J.; Gale, M. Human Immunodeficiency Virus Type 1 Mediates Global Disruption of Innate Antiviral Signaling and Immune Defenses within Infected Cells. J. Virol. 2009, 83, 10395–10405. [Google Scholar] [CrossRef] [Green Version]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Marno, K.M.; Ogunkolade, B.W.; Pade, C.; Oliveira, N.M.; O’Sullivan, E.; McKnight, A. Novel restriction factor RNA-associated early-stage anti-viral factor (REAF) inhibits human and simian immunodeficiency viruses. Retrovirology 2014, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.; Sumner, R.P.; Rasaiyaah, J.; Tan, C.P.; Rodriguez-Plata, M.T.; Van Tulleken, C.; Fink, D.; Zuliani-Alvarez, L.; Thorne, L.; Stirling, D.; et al. HIV-1 Vpr antagonizes innate immune activation by targeting karyopherin-mediated NF-kappaB/IRF3 nuclear transport. Elife 2020, 9, e60821. [Google Scholar] [CrossRef] [PubMed]
- Azimi, F.C.; Lee, J.E. Structural perspectives on HIV-1 Vif and APOBEC3 restriction factor interactions. Protein Sci. 2019, 29, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Chaipan, C.; Smith, J.L.; Hu, W.-S.; Pathak, V.K. APOBEC3G Restricts HIV-1 to a Greater Extent than APOBEC3F and APOBEC3DE in Human Primary CD4+ T Cells and Macrophages. J. Virol. 2012, 87, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Kajaste-Rudnitski, A.; Coradin, T.; Saba, E.; Della Chiara, G.; Barbagallo, M.; Graziano, F.; Alfano, M.; Cassol, E.; Vicenzi, E.; et al. M1 polarization of human monocyte-derived macrophages restricts pre and postintegration steps of HIV-1 replication. AIDS 2013, 27, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Greenwell-Wild, T.; Nares, S.; Jin, W.; Lei, K.J.; Rangel, Z.G.; Munson, P.J.; Wahl, S.M. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 2007, 110, 393–400. [Google Scholar] [CrossRef]
- Berger, G.; Durand, S.; Fargier, G.; Nguyen, X.-N.; Cordeil, S.; Bouaziz, S.; Muriaux, D.; Darlix, J.-L.; Cimarelli, A. APOBEC3A Is a Specific Inhibitor of the Early Phases of HIV-1 Infection in Myeloid Cells. PLoS Pathog. 2011, 7, e1002221. [Google Scholar] [CrossRef] [Green Version]
- Covino, D.A.; Kaczor-Urbanowicz, K.E.; Lu, J.; Chiantore, M.V.; Fiorucci, G.; Vescio, M.F.; Catapano, L.; Purificato, C.; Galluzzo, C.M.; Amici, R.; et al. Transcriptome Profiling of Human Monocyte-Derived Macrophages Upon CCL2 Neutralization Reveals an Association Between Activation of Innate Immune Pathways and Restriction of HIV-1 Gene Expression. Front. Immunol. 2020, 11, 2129. [Google Scholar] [CrossRef] [PubMed]
- Akimova, E.; Gassner, F.J.; Schubert, M.; Rebhandl, S.; Arzt, C.; Rauscher, S.; Tober, V.; Zaborsky, N.; Greil, R.; Geisberger, R. SAMHD1 restrains aberrant nucleotide insertions at repair junctions generated by DNA end joining. Nucleic Acids Res. 2021, 49, 2598–2608. [Google Scholar] [CrossRef]
- Wu, L. SAMHD1: A new contributor to HIV-1 restriction in resting CD4+T-cells. Retrovirology 2012, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woelk, C.H.; Ottones, F.; Plotkin, C.R.; Du, P.; Royer, C.D.; Rought, S.E.; Lozach, J.; Sasik, R.; Kornbluth, R.S.; Richman, D.D.; et al. Interferon gene expression following HIV type 1 infection of monocyte-derived macrophages. AIDS Res. Hum. Retrovir. 2004, 20, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Nasr, N.; Maddocks, S.; Turville, S.G.; Harman, A.N.; Woolger, N.; Helbig, K.J.; Wilkinson, J.; Bye, C.R.; Wright, T.K.; Rambukwelle, D.; et al. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 2012, 120, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Taylor, K.; Ciechonska, M.; Collins, S.E.; Duncan, R.; Mossman, K.L. Membrane Perturbation Elicits an IRF3-Dependent, Interferon-Independent Antiviral Response. J. Virol. 2011, 85, 10926–10931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, C.; Jensen, S.B.; Jakobsen, M.R.; Cheshenko, N.; Horan, K.A.; Moeller, H.B.; Gonzalez-Dosal, R.; Rasmussen, S.B.; Christensen, M.H.; O Yarovinsky, T.; et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 2012, 13, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Ducroux, A.; Ponnurangam, A.; Vieyres, G.; Franz, S.; Musken, M.; Zillinger, T.; Malassa, A.; Ewald, E.; Hornung, V.; et al. cGAS-Mediated Innate Immunity Spreads Intercellularly through HIV-1 Env-Induced Membrane Fusion Sites. Cell Host Microbe 2016, 20, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, V.; Ruffin, N.; San-Roman, M.; Benaroch, P. Myeloid Cell Interaction with HIV: A Complex Relationship. Front. Immunol. 2017, 8, 1698. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The Interferon-Inducible MxB Protein Inhibits HIV-1 Infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef] [Green Version]
- A Matreyek, K.; Wang, W.; Serrao, E.; Singh, P.K.; Levin, H.L.; Engelman, A. Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology 2014, 11, 90. [Google Scholar] [CrossRef] [Green Version]
- Buffone, C.; Kutzner, J.; Opp, S.; Martinez-Lopez, A.; Selyutina, A.; Coggings, S.A.; Studdard, L.R.; Ding, L.; Kim, B.; Spearman, P.; et al. The ability of SAMHD1 to block HIV-1 but not SIV requires expression of MxB. Virology 2019, 531, 260–268. [Google Scholar] [CrossRef]
- Bhargava, A.; Lahaye, X.; Manel, N. Let me in: Control of HIV nuclear entry at the nuclear envelope. Cytokine Growth Factor Rev. 2018, 40, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Giese, S.; Marsh, M. Tetherin Can Restrict Cell-Free and Cell-Cell Transmission of HIV from Primary Macrophages to T Cells. PLoS Pathog. 2014, 10, e1004189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhl, B.D.; Sloan, R.D.; Donahue, D.A.; Liang, C.; Wainberg, M.A. Vpu-mediated tetherin antagonism of ongoing HIV-1 infection in CD4+ T-cells is not directly related to the extent of tetherin cell surface downmodulation. Virology 2011, 417, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Pan, Q.; Ding, S.; Rong, L.; Liu, S.-L.; Geng, Y.; Qiao, W.; Liang, C. The N-Terminal Region of IFITM3 Modulates Its Antiviral Activity by Regulating IFITM3 Cellular Localization. J. Virol. 2012, 86, 13697–13707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Pierini, V.; Gallucci, L.; Stürzel, C.M.; Kirchhoff, F.; Fackler, O.T. SERINC5 Can Enhance Proinflammatory Cytokine Production by Primary Human Myeloid Cells in Response to Challenge with HIV-1 Particles. J. Virol. 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Zutz, A.; Schölz, C.; Schneider, S.; Pierini, V.; Münchhoff, M.; Sutter, K.; Wittmann, G.; Dittmer, U.; Draenert, R.; Bogner, J.R.; et al. SERINC5 Is an Unconventional HIV Restriction Factor That Is Upregulated during Myeloid Cell Differentiation. J. Innate Immun. 2020, 12, 399–409. [Google Scholar] [CrossRef]
- Guo, J.; Wu, Y. Chemokine Receptor CCR5 Antagonist Maraviroc: Medicinal Chemistry and Clinical Applications. Curr. Top. Med. Chem. 2014, 14, 1504–1514. [Google Scholar] [CrossRef] [Green Version]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 Env on SERINC5 Antagonism. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo-Pereira, J.M.; Canhão, P.; Calado, M.; Santos-Costa, Q.; Barroca, P. Inhibition of HIV Replication by Host Cellular Factors. In Trends in Basic and Therapeutic Options in HIV Infection—Towards a Functional Cure; IntechOpen: London, UK, 2015. [Google Scholar]
- Saulle, I.; Ibba, S.V.; Vittori, C.; Fenizia, C.; Mercurio, V.; Vichi, F.; Caputo, S.L.; Trabattoni, D.; Clerici, M.; Biasin, M. Sterol metabolism modulates susceptibility to HIV-1 Infection. AIDS 2020, 34, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.L.; Poli, G.; Fox, L.; Hardy, E.; Fauci, A.S. HIV replication in IL-2-stimulated peripheral blood mononuclear cells is driven in an autocrine/paracrine manner by endogenous cytokines. J. Immunol. 1995, 154. [Google Scholar]
- Wu, T.; Ma, F.; Ma, X.; Jia, W.; Pan, E.; Cheng, G.; Chen, L.; Sun, C. Regulating Innate and Adaptive Immunity for Controlling SIV Infection by 25-Hydroxycholesterol. Front. Immunol. 2018, 9, 2686. [Google Scholar] [CrossRef]
- Turner, J.; Torrelles, J.B. Mannose-capped lipoarabinomannan in Mycobacterium tuberculosis pathogenesis. Pathog. Dis. 2018, 76, fty026. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.-S.; Hsieh, S.-L. CLEC2 and CLEC5A: Pathogenic Host Factors in Acute Viral Infections. Front. Immunol. 2019, 10, 2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassol, E.; Cassetta, L.; Rizzi, C.; Gabuzda, D.; Alfano, M.; Poli, G. Dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin mediates HIV-1 infection of and transmission by M2a-polarized macrophages in vitro. AIDS 2013, 27, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicala, C.; Arthos, J.; Fauci, A.S. Role of T-cell trafficking in the pathogenesis of HIV disease. Curr. Opin. HIV AIDS 2019, 14, 115–120. [Google Scholar] [CrossRef]
- Richter, S.N.; Frasson, I.; Palu, G. Strategies for inhibiting function of HIV-1 accessory proteins: A necessary route to AIDS therapy? Curr. Med. Chem. 2009, 16, 267–286. [Google Scholar] [CrossRef] [Green Version]
- Mori, L.; Valente, S.T. Cure and Long-Term Remission Strategies. Methods Mol. Biol. 2022, 2407, 391–428. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, X.; De Clercq, E. Cessation of HIV-1 transcription by inhibiting regulatory protein Rev-mediated RNA transport. Curr. HIV Res. 2009, 7, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Dekaban, G.A.; Dikeakos, J.D. HIV-I Nef inhibitors: A novel class of HIV-specific immune adjuvants in support of a cure. AIDS Res. Ther. 2017, 14, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dong, X.; Zhang, W.; Chen, T.; Yu, B.; Zhao, W.; Yang, Y.; Wang, X.; Hu, Q.; Wang, X. High-Throughput NanoBiT-Based Screening for Inhibitors of HIV-1 Vpu and Host BST-2 Protein Interaction. Int. J. Mol. Sci. 2021, 22, 9308. [Google Scholar] [CrossRef]
- Ikeda, T.; Yue, Y.; Shimizu, R.; Nasser, H. Potential Utilization of APOBEC3-Mediated Mutagenesis for an HIV-1 Functional Cure. Front. Microbiol. 2021, 12, 686357. [Google Scholar] [CrossRef] [PubMed]
- González, M.E. The HIV-1 Vpr Protein: A Multifaceted Target for Therapeutic Intervention. Int. J. Mol. Sci. 2017, 18, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, R.E.; Dong, S.X.M.; Gajanayaka, N.; Ali, H.; Cassol, E.; Cameron, W.D.; Korneluk, R.; Tremblay, M.J.; Angel, J.B.; Kumar, A. Role of RIPK1 in SMAC mimetics-induced apoptosis in primary human HIV-infected macrophages. Sci. Rep. 2021, 11, 22901. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vidal, E.; Badia, R.; Pujantell, M.; Castellví, M.; Felip, E.; Clotet, B.; Riveira-Muñoz, E.; Ballana, E.; Esté, J.A. Dual effect of the broad spectrum kinase inhibitor midostaurin in acute and latent HIV-1 infection. Antivir. Res. 2019, 168, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassol, E.; Cassetta, L.; Rizzi, C.; Alfano, M.; Poli, G. M1 and M2a Polarization of Human Monocyte-Derived Macrophages Inhibits HIV-1 Replication by Distinct Mechanisms. J. Immunol. 2009, 182, 6237–6246. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Buckley, M.; Britton, B.; Mu, Y.; Warner, K.; Kumar, S.; Cory, T.J. Polarized macrophage subsets differentially express the drug efflux transporters MRP1 and BCRP, resulting in altered HIV production. Antivir. Chem. Chemother. 2018, 26. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassol, E.; Cassetta, L.; Alfano, M.; Poli, G. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 2009, 87, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, E.; Rochat, M.-A.; Duo, L.; Speck, R.F. Triggering TLR2, -3, -4, -5, and -8 Reinforces the Restrictive Nature of M1- and M2-Polarized Macrophages to HIV. J. Virol. 2014, 88, 9769–9781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrini, F.F.; Marelli, S.S.; A Kajaste-Rudnitski, A.; Lusic, M.M.; Van Lint, C.; Das, A.T.; Harwig, A.; Berkhout, B.; Vicenzi, E. HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology 2015, 12, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddine, F.B.N.; Forlani, G.; Lombardo, L.; Tedeschi, A.; Tosi, G.; Accolla, R.S. CIITA-driven MHC class II expressing tumor cells can efficiently prime naive CD4+ TH cells in vivo and vaccinate the host against parental MHC-II-negative tumor cells. OncoImmunology 2016, 6, e1261777. [Google Scholar] [CrossRef] [Green Version]
- Viglianti, G.A.; Planelles, V.; Hanley, T.M. Interactions with Commensal and Pathogenic Bacteria Induce HIV-1 Latency in Macrophages through Altered Transcription Factor Recruitment to the LTR. J. Virol. 2021, 95, e02141-20. [Google Scholar] [CrossRef]
- Robinson, T.O.; Zhang, M.; Ochsenbauer, C.; Smythies, L.E.; Cron, R.Q. CD4 regulatory T cells augment HIV-1 expression of polarized M1 and M2 monocyte derived macrophages. Virology 2017, 504, 79–87. [Google Scholar] [CrossRef]
- Wong, M.E.; Johnson, C.J.; Hearps, A.C.; Jaworowski, A. Development of a Novel In Vitro Primary Human Monocyte-Derived Macrophage Model To Study Reactivation of HIV-1 Transcription. J. Virol. 2021, 95, JVI0022721. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- El Costa, H.; Quillay, H.; Marlin, R.; Cannou, C.; Duriez, M.; Benjelloun, F.; de Truchis, C.; Rahmati, M.; Ighil, J.; Barré-Sinoussi, F.; et al. The local environment orchestrates mucosal decidual macrophage differentiation and substantially inhibits HIV-1 replication. Mucosal Immunol. 2016, 9, 634–646. [Google Scholar] [CrossRef] [Green Version]



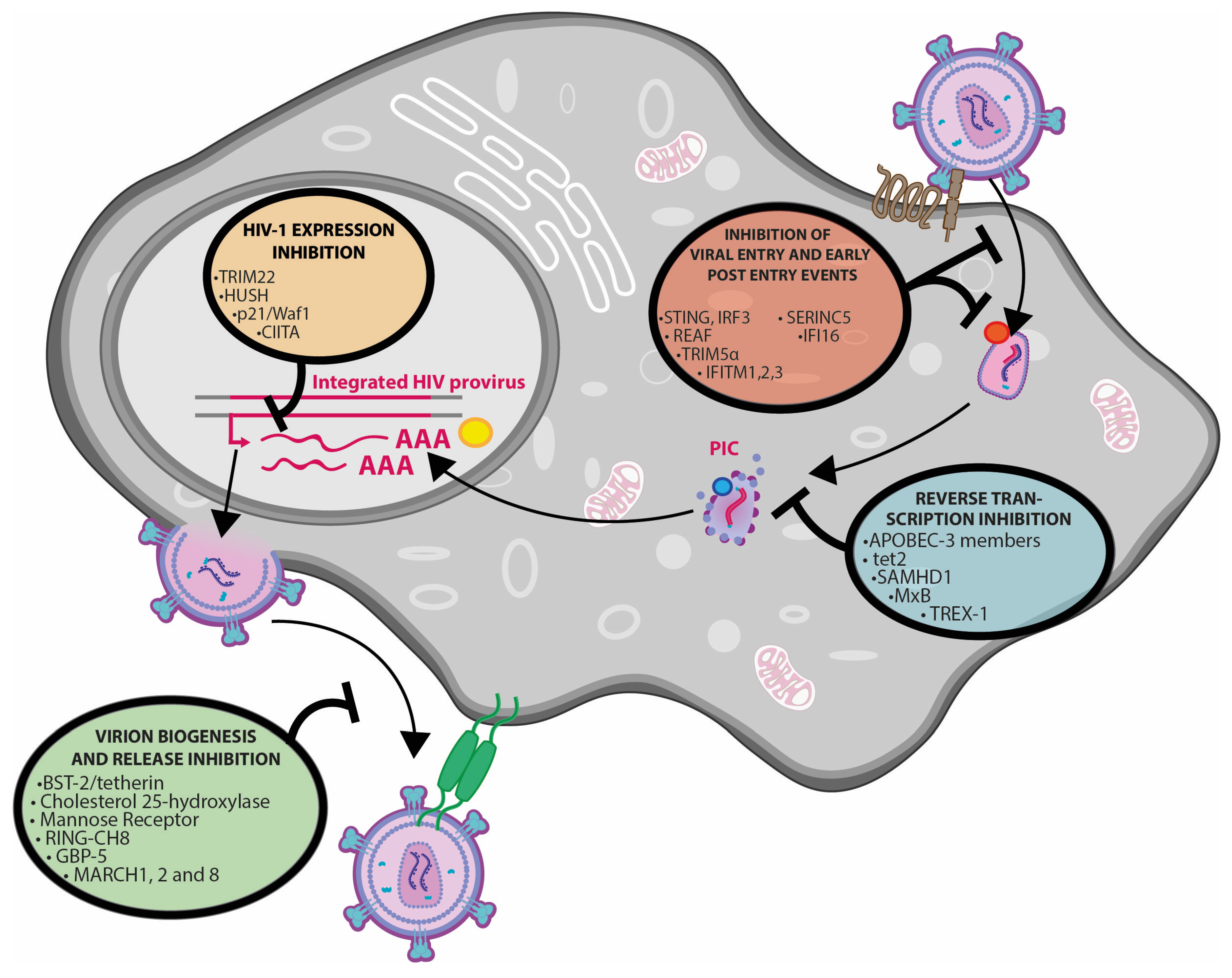{kind=link}
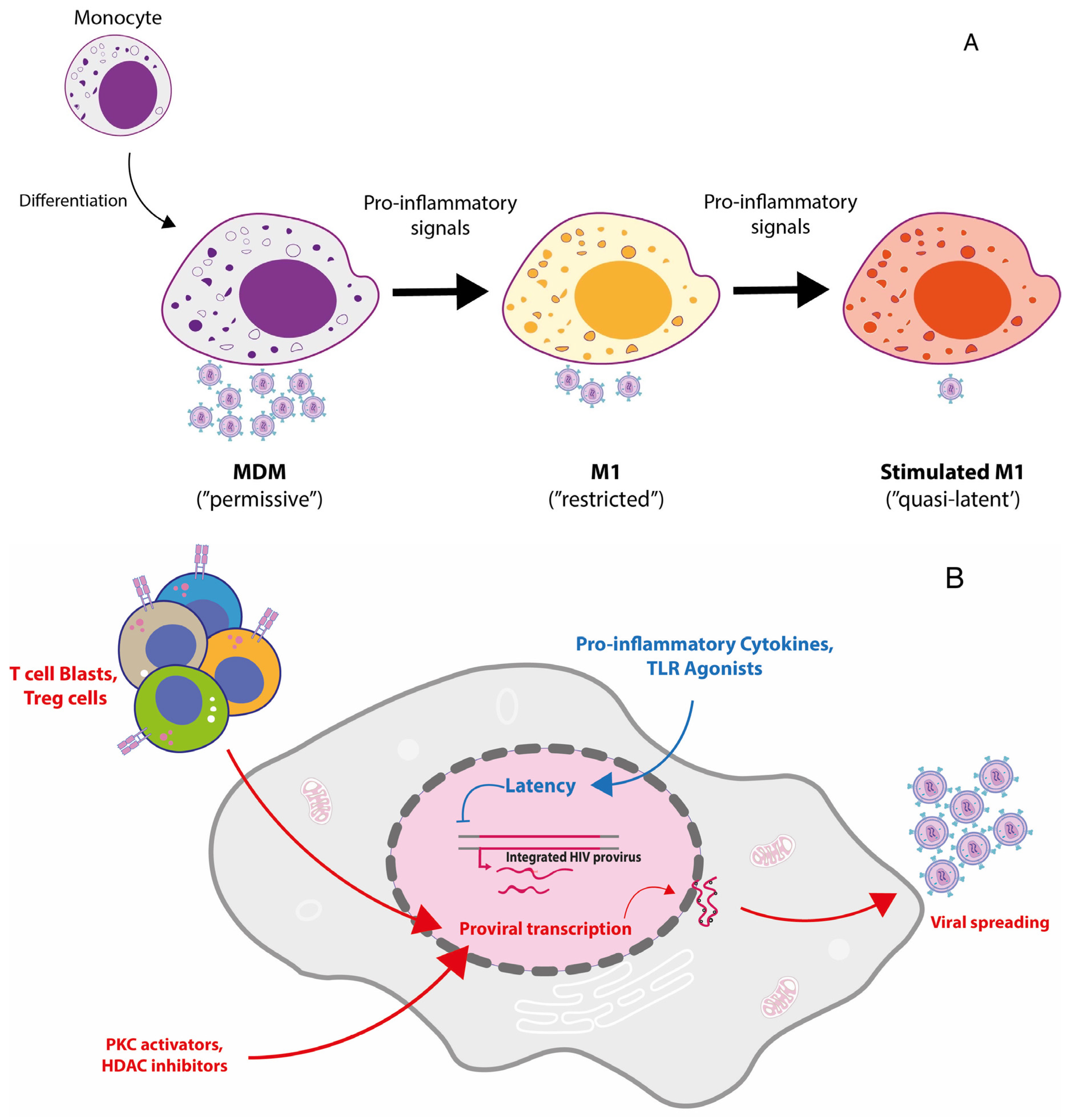{kind=link}
| CD4+ T Cells | Macrophages | Refs. | Notes | |
|---|---|---|---|---|
| Entry receptors | CD4, CCR5, CXCR4 | CD4, CCR5, CXCR4 | [2] | Although macrophages express CXCR4 productive infection is usually associated with HIV CCR5 use |
| Cell proliferation | Yes | No | [12,13] | |
| Cytopathic effect, cell depletion in vitro | Yes | No | [9] | |
| Cytopathic effect, cell depletion in vivo | Yes | No | [2,9] | CD4 T cell depletion in vivo is likely the result of different processes in addition to virus-induced cytopathicity |
| Main pathogenetic consequence | Profound immunodeficiency, opportunistic infections, cancer | Tissue pathology, brain infection (encephalitis) | [2,14] | |
| Virus budding and release | Plasma membrane only | Plasma membrane and VCC | [15,16] | VCC are defined as invaginations of the plasma membrane connected or not to the cell surface and the extracellular environment |
| Role as viral reservoirs in cART-treated individuals | Well-demonstrated in the case of latently infected “resting memory” cells | Strong evidence in support of TRM | [17,18,19,20] | TRM are credited with a longer ½ life than MDM |
| Restriction Factor | HIV Life Cycle Step Affected | Mechanism of Action | Counteracting Viral Protein | Key Refs | Notes |
|---|---|---|---|---|---|
| SERINC3/5 | Viral entry | Prevention of virion-cell fusion | Nef | [25,26] | |
| IFITM1, 2, 3 | Viral entry | Incorporation into nascent HIV-1 virions and prevention of cell fusion | Vpr | [27] | |
| CH25H | Viral entry | Prevention of virion-cell fusion | [28,29] | ||
| STING | Post-entry events | Induction of the IFN response | Vpr, Vpx | [30,31,32] | HIV-2 only |
| REAF | Early post-entry events | Unclear/unknown | Vpr | [33] | |
| TRIM5α | Early post-entry events | Degradation of the incoming viral capsid | [23,34,35,36,37] | Human TRIM5α prevents animal lentivirus infection, whereas cyclophilin A prevents its binding to HIV in human cells | |
| APOBEC3 members | Reverse transcription | C to A hypermutation | Vif | [23] | |
| Tet2 | Reverse transcription | Cytosine demethylation | Vpr | [38] | |
| SAMHD1 | Reverse transcription | Depletion of dNTP pool | Vpx | [39,40] | SAMHD1 gene is involved in the Aicardi Goutières Syndrome |
| TREX-1 | Reverse transcription | prevention of IFN induction | [41,42] | TREX1 gene is involved in the Aicardi Goutières Syndrome | |
| IFI16* | Reverse transcription | Induction of IFN response | [43] | IFN-inducible protein 16 interacts with single stranded HIV DNA | |
| Mx2/MxB | Post-reverse transcription | Interaction with PIC | [44,45,46] | PIC: Pre-Integration Complex | |
| TRIM22 | Integrated provirus | Transcriptional repression | [47,48] | Inhibition mediated by interference with Sp1 | |
| NF-kB1 (p50) homodimers | Integrated provirus | Transcriptional repression | [49,50] | NF-kB1 can form heterodimers with C-terminally truncated STAT5 to repress proviral transcription | |
| p21/Waf1 | Integrated provirus | Transcriptional repression | [51,52] | ||
| CIITA | Integrated provirus | Transcriptional repression | [53,54] | Class II transactivator, also repressed HTLV-1/2 Tax transcriptional activity | |
| HUSH Complex | Integrated provirus | Transcriptional repression | Vpx, Vpr | [55,56] | HUman Silencing Hub |
| BST-2/Tetherin | Budding and virion release | Prevention of virion release from plasma membrane | Vpu (Nef) | [57,58,59,60] | IFN-α stimulation upregulates BST-2/tetherin expression. In addition, Tetherin can trigger NF-kB activation after binding of Vpu-defective HIV |
| GBP-5 | Budding and virion release | Prevention of envelope incorporation into virions | Vpu | [61] | |
| MARCH1, 2 and 8 | Budding and virion release | Prevention of envelope incorporation into virions | [62,63,64] | ||
| Mannose Receptor | Budding and virion release | Prevention of envelope incorporation into virions | VpR, Nef | [65,66] |
| HIV Protein | Correlated RF | Pharmacologic Inhibitor | Key References |
|---|---|---|---|
| Tat | n.a. | Didehydro-Cortistatin A | [121] |
| Rev | n.a. | Several molecules | [122] |
| Nef | SerinC3/C5 | Several molecules | [123] |
| Vpu | BST-2/Tetherin | Several molecules | [124] |
| Vif | APOBEC family members | [125] | |
| Vpr | Several (see Table 1) | [126,127] | |
| Vpx | SAMHD1 | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagani, I.; Demela, P.; Ghezzi, S.; Vicenzi, E.; Pizzato, M.; Poli, G. Host Restriction Factors Modulating HIV Latency and Replication in Macrophages. Int. J. Mol. Sci. 2022, 23, 3021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063021
Pagani I, Demela P, Ghezzi S, Vicenzi E, Pizzato M, Poli G. Host Restriction Factors Modulating HIV Latency and Replication in Macrophages. International Journal of Molecular Sciences. 2022; 23(6):3021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063021
Chicago/Turabian StylePagani, Isabel, Pietro Demela, Silvia Ghezzi, Elisa Vicenzi, Massimo Pizzato, and Guido Poli. 2022. "Host Restriction Factors Modulating HIV Latency and Replication in Macrophages" International Journal of Molecular Sciences 23, no. 6: 3021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063021