
The Signaling Pathway of TNF Receptors: Linking Animal Models of Renal Disease to Human CKD


Abstract
:1. Chronic Kidney Disease—A Public Health Issue
2. Inflammation as an Essential Component of CKD
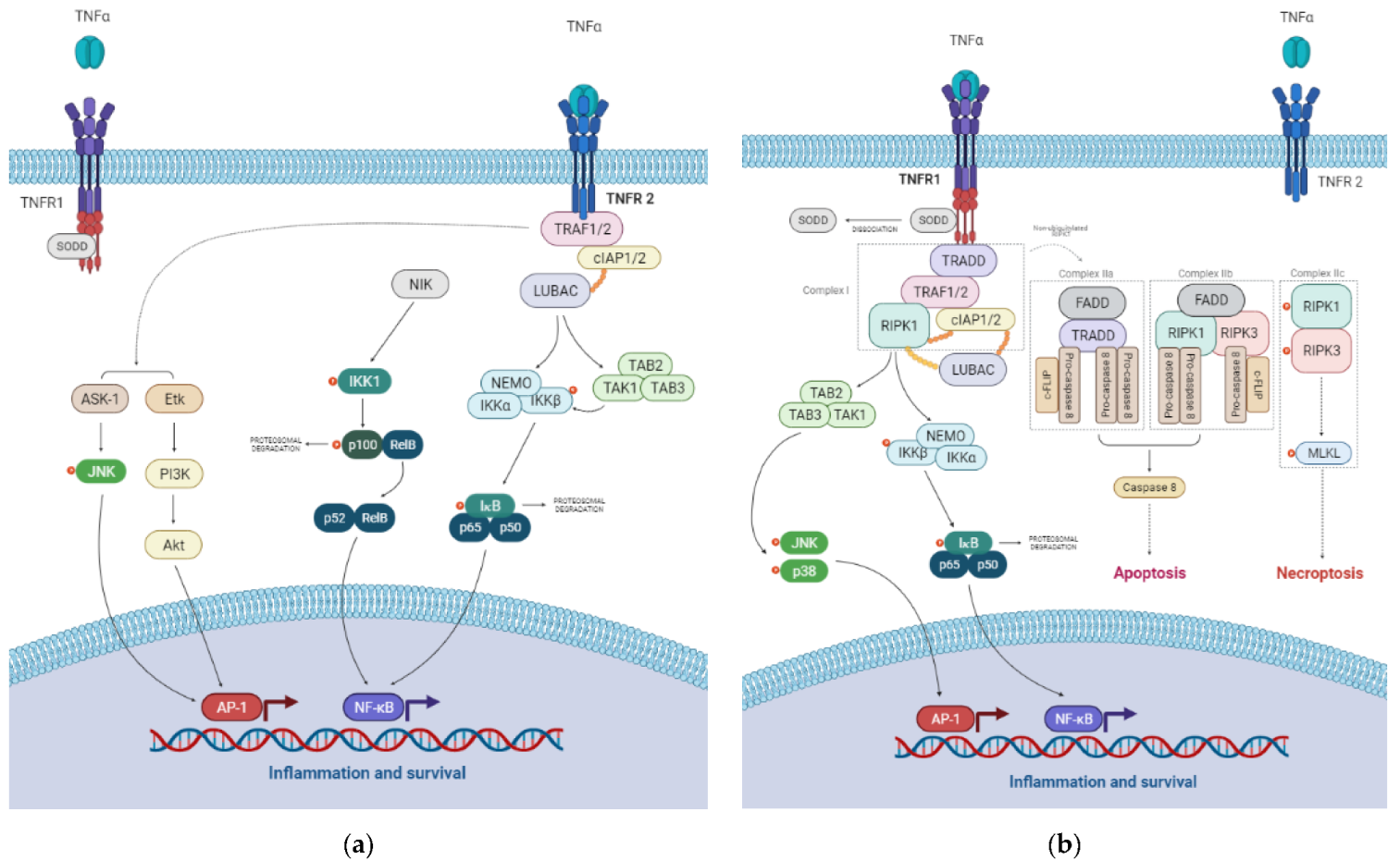
3. The TNF Signaling Pathway
3.1. TNFR1 Signaling Pathways
3.2. TNFR2 Signaling Pathways
4. Involvement of TNF Receptors on Renal Deterioration
4.1. Studies Addressing TNF-α and TNFRs in Animal Models

{kind=link}
| Year | Study Model | Methods | Study Outcomes | Reference |
|---|---|---|---|---|
| 1989 | Anti GBM nephritis rat model | Pretreatment with human TNF-α | Pretreatment of rats with TNF-α increased the glomerular neutrophil influx and exacerbated glomerular injury, judged by the increased albuminuria and the prevalence of glomerular capillary thrombi. | [83] |
| 1998 | Anti-GBM nephritis mice model | Tnf-α knockout mice | In TNF-deficient mice, the influx of lymphocytes was reduced, the development of proteinuria was delayed and the formation of crescents was almost completely prevented. | [87] |
| 1999 | UUO mice model | Tnfr1 and Tnfr2 knockout | Individual knockout of the TNFRs genes resulted in significantly less NF-kB activation compared with the WT. Tnfr1 knockout showed a significant reduction in Tnf-α mRNA levels compared with WT or Tnfr2 knockout mice. | [93] |
| 2001 | Rat model of crescentic glomerulonephritis | TNF-α blockade with sTNFR1 | Treatment with sTNFR1 caused a marked reduction in albuminuria, reduced glomerular cell infiltration, activation, and proliferation, and prevented the development of crescents. | [88] |
| 2003 | Mice model of cisplatin-induced acute renal failure | Tnfr1 and Tnfr2 knockout | Tnfr2-deficient mice developed less-severe renal dysfunction and showed reduced necrosis, apoptosis, and leukocyte infiltration into the kidney, and lower renal and serum TNF levels compared with either Tnfr1-deficient or WT mice. | [94] |
| 2003 | Streptozotocin (STZ)-induced diabetic rats | Administration of a TNF antagonist (TNFR:Fc) | Administration of a TNF antagonist reduces urinary TNF-α excretion and prevents sodium retention and renal hypertrophy. TNF-α contributes to early diabetic nephropathy, and its inhibition may attenuate early pathological changes. | [98] |
| 2005 | Rat model of nephrotoxic nephritis | Administration of anti-TNF-α antibody | Neutralization of endogenous TNF-α reduces glomerular inflammation, crescent formation, and tubulointerstitial scarring, with preservation of renal function. | [103] |
| 2005 | Anti-GBM nephritis mice model | Tnfr1 or Tnfr2 knockout | Lack of Tnfr1 resulted in excessive renal T cell accumulation and an associated reduction in apoptosis of these cells. Tnfr2-deficient mice were completely protected from glomerulonephritis, despite an intact systemic immune response. | [92] |
| 2005 | UUO rat model | TNF-α blockade with PEG-sTNFR1 | Treatment with PEG-sTNFR1 reduced tissue Tnf-α and protein production, renal tubular cell apoptosis, and caspase activity. | [89] |
| 2007 | UUO rat model | TNF-α blockade with PEG-sTNFR1 | Renal obstruction induced increased tissue TNF-α and several markers of renal fibrosis, whereas treatment with PEG-sTNFR1 significantly reduced each of these markers of renal fibrosis. | [90] |
| 2007 | Rat model of kidney transplantation | Treatment with cyclsporine | In rats with acute allograft rejection, significantly elevated expression of TNFR2 was observed in tubular epithelial cells, podocytes, B cells, and monocytes/macrophages. TNFR2 expression levels were associated with renal function. | [104] |
| 2007 | STZ-induced diabetic rats | Administration TNF-α inhibitors, Infliximab and FR167653 | TNF-α inhibition with infliximab and FR167653 decreased urinary albumin excretion, suggesting the role of TNF-α in the pathogenesis of diabetic nephropathy, with TNF-α inhibition is a potential therapeutic strategy. | [100] |
| 2008 | UUO mice model | Tnf-α knockout | Tnf-deficient mice showed an increase of extracellular matrix in the kidneys and infiltrating macrophages, explained by the increased TNFR2 expression level. | [105] |
| 2009 | SLE prone mice models | Tnfr1, Tnfr2 and double Tnfr1/2 knockout | Doubly-deficient mice developed accelerated pathological and clinical nephritis, while mice deficient in either TNFR, alone, did not differ from each other or from WT controls. | [106] |
| 2010 | ANG II-dependent mice model of hypertension | Tnfr1 knockout | Angiotensin II inhibited renal Tnfr1 mRNA accumulation, while increasing that of Tnfr2. Deletion of Tnfr1 was associated with increased albuminuria and creatinine clearance, in response to ANG II infusion. | [91] |
| 2013 | Anti-GBM nephritis mice model | Tnf-α, Tnfr1 and Tnfr2 knockout | Tnfr2 deficiency resulted in a reduction in renal macrophage but not neutrophil accumulation, while Tnfr1 deletion prevented the influx of both leukocyte subsets. | [79] |
| 2013 | TNF-induced inflammation mice model | Tnfr1, Tnfr2 and double Tnfr1/2 knockout | TNF-induced glomerular leukocyte infiltration was abrogated in Tnfr1-deficient mice, whereas Tnfr2-deficiency decreased mononuclear phagocytes infiltrates, but not neutrophils. | [107] |
| 2014 | Mice models of LPS- or TNF-induced acute endotoxemia | Tnfr1 knockout | LPS and TNF-treated WT models showed alterations of glomerular endothelium, increased albuminuria, and decreased GFR. The effects of LPS on the glomerular endothelial surface layer, GFR, and albuminuria were diminished in Tnfr1 knockout mice. | [108] |
| 2014 | Type 2 diabetic model of the KK-Ay mouse | TNF-α inhibition with Etanercept (ETN) | Renal mRNA and/or protein levels of Tnfr2, but not Tnf-α and Tnfr1, in ETN-treated mice were significantly decreased. ETN may exert a renal protective effect via inhibition of the inflammatory pathway activated by TNFR2 rather than TNFR1. | [99] |
| 2017 | Mice with CaOx nephrocalcinosis-related CKD | Tnfr1, Tnfr2 and double Tnfr1/2 knockout | WT mice developed progressive CKD, while Tnfr1-, Tnfr2-, and Tnfr1/2-deficient mice lacked intrarenal CaOx deposition and tubular damage, despite exhibiting similar levels of hyperoxaluria. | [95] |
| 2019 | STZ-induced diabetic rats | Treatment with adalimumabe, a TNF-α inhibitor | TNF-α inhibition reduced albuminuria, glomerular injury, and tubular injury in STZ-induced diabetic rats. TNF-α inhibition reduced the NLRP3 inflammasome in tubules and decreased expression of tubular IL-6 and IL-17A mRNA. | [97] |
| 2020 | Rodent models of 2,8-DHA crystal nephropathy | Tnfr1 and Tnfr2 knockout | Deletion of Tnfr1 significantly reduced tubular inflammation, thereby ameliorating the disease course. In contrast, genetic deletion of Tnfr2 had no effect on the manifestations of 2,8-DHA nephropathy. | [96] |
| 2021 | Ischemia-reperfusion mice model | Clamping of the renal pedicles | Proximal tubular cells exhibited a profibrotic and proinflammatory profile, and a marked transcriptional activation of NF-κB and AP-1 signaling pathways. | [102] |
4.2. Studies Addressing TNF-α and TNFRs in Human Kidney Disease and Related Clinical Outcomes
4.3. Anti-TNF-α Theraphy in Patients with Impaired Kidney Function
| Year | Study Type | Study Population | Biomarkers | Study Outcomes | Reference |
|---|---|---|---|---|---|
| 1994 | Cross-sectional | 26 non-HD CKD patients, 61 HD patients, 43 renal transplant recipients and 34 healthy controls | Serum levels of TNFR? | All patient groups showed significantly higher TNFR levels compared to the control group. A correlation of TNFR and creatinine levels was only found in the group of non-dialyzed CKD patients. | [109] |
| 2005 | Retrospective cohort | 687 individuals from the CARE trial study, with CKD and previous myocardial infarction | Serum levels of TNFR2 | Higher TNFR2 is independently associated with faster rates of kidney function loss in CKD. Inflammation may mediate the loss of kidney function among subjects with CKD and concomitant coronary disease. | [110] |
| 2007 | Cross-sectional | 38 patients with SLE and 15 healthy controls | Urinary levels of TNFR1 | Urinary TNFR1 levels were elevated in patients with lupus nephritis and correlated with proteinuria and SLE disease activity index scores. | [117] |
| 2007 | Prospective cohort | 3075 adults aged 70 to 79 | Serum levels of TNF-α, TNFR1 and TNFR2 | In an elderly cohort of patients with eGFR ≥ 60 mL/min/1.73 m2, cystatin C was strongly associated with TNF-α and the TNFRs. | [144] |
| 2008 | Cross-sectional | 6814 participants free of cardiovascular disease, from the MESA study | Circulating levels of TNFR1 | Creatinine-based eGFR had significant correlations with TNFR1, in both participants with and without CKD. | [145] |
| 2009 | Cross-sectional | 96 human renal allograft biopsies | Renal TNFR2 expression | In human renal transplant biopsies, there was an increase in the number of TNFR2-positive podocytes, in tubular epithelial cells, B cells, and monocytes/macrophages. | [104] |
| 2009 | Cross-sectional | 667 participants with diabetes | Serum levels of TNF-α, TNFR1 and TNFR2 | Elevated concentrations of serum markers of the TNF-α pathway were strongly associated with decreased renal function in T1D patients without proteinuria. | [146] |
| 2010 | Prospective cohort | 55 patients with biopsy-proven primary glomerulonephritis and 20 healthy controls | Urinary levels of TNFR1 | Elevated TNFR1 urinary levels predicted renal function decline and advanced renal interstitial fibrosis in patients with primary nephropathy. | [147] |
| 2010 | Cross-sectional | 3294 participants from the Framingham Offspring Study, 291 of them with CKD | Serum levels of TNF-α and TNFR2 | A significant proportion of variability in TNFR2 concentration was explained by CKD status and higher cystatin C quartiles. Higher concentrations of TNF and TNFR2 were associated with CKD status, higher cystatin C, and higher UACR. | [148] |
| 2011 | Prospective cohort | 4926 patients followed for 15 years | Serum levels of TNFR2 | For the risk of developing incident CKD among those who were CKD-free at baseline, only TNFR2 and IL-6 levels, but not CRP, were positively associated with incident CKD. | [149] |
| 2012 | Prospective cohort | 3939 participants with established CKD | Plasma levels of TNF-α | Biomarkers of inflammation (cytokines and acute phase proteins) were higher in participants with lower levels of kidney function and higher levels of albuminuria. | [9] |
| 2012 | Prospective cohort | 628 patients with T1D, normal renal function, and no proteint2uria | Serum levels of TNFR1 and TNFR2 | Elevated serum concentrations of TNFR1 and TNFR2 were strongly associated with early renal function loss, progression to CKD stage 3 or higher, in patients with T1D who had normal renal function. | [120] |
| 2012 | Prospective cohort | 410 patients with T2D | Serum levels of TNFR1 and TNFR2 | Elevated concentrations of circulating TNFRs in patients withT2D at baseline were very strong predictors of the subsequent progression to ESRD in subjects with and without proteinuria. | [121] |
| 2012 | Prospective cohort | 12 patients with active lupus nephritis, 14 with inactive SLE, and 14 healthy subjects | Serum levels of TNF-α and TNFR2 | TNFR2 serum levels were elevated in all patients with active lupus nephritis and declined after clinical remission. | [118] |
| 2013 | Prospective cohort | 84 glomerulonephritis patients under immunosuppressive therapy and 18 healthy controls | Serum and urine levels of TNFR1 and TNFR2 | Urinary levels, but not serum levels, of TNFR1 and TNFR2 were effective in predicting a favorable response to immunosuppressive treatment in patients with primary glomerulonephritis. | [150] |
| 2014 | Prospective cohort | Patients with T1D and normoalbuminuria (286) or microalbuminuria (248) | Serum levels of TNF-α, TNFR1 and TNFR2 | In both groups, the strongest determinants of renal decline were baseline serum concentrations of uric acid and TNFRs. Renal decline was not associated with sex or baseline serum concentration of the other measured markers. | [122] |
| 2014 | Prospective cohort | 113 patients with biopsy-proven iMN and 43 healthy volunteers | Serum levels of TNFR1 and TNFR2 | Estimated glomerular filtration rate and proteinuria tended to worsen as the TNFRs levels increased. Renal tubular TNFRs expression was associated with circulating TNFRs levels. | [115] |
| 2014 | Prospective cohort | 522 T2D patients with DKD | Serum levels of TNFR1 | TNFR1 is a strong prognostic factor for all-cause mortality in T2D with renal dysfunction, and its clinical utility is suggested in addition to established risk factors for all-cause mortality. | [130] |
| 2014 | Prospective cohort | 429 patients with T1D and overt nephropathy | Plasma levels of TNFR1 | Circulating levels of TNFR1 were highly correlated with eGFR, especially in patients with an eGFR < 60 mL/min/1.73 m2. Circulating levels of the TNFR1 also remained associated with ESRD after adjusting for the competing risk of death. | [151] |
| 2014 | Prospective cohort | 349 T1D patients with proteinuria and CKD staged 1–3 | Serum levels of TNFR2 | Serum TNFR2 was the strongest determinant of renal decline and ESRD risk. The rate of eGFR loss became steeper with rising concentration of TNFR2, and elevated HbA1c augmented the strength of this association. | [152] |
| 2015 | Prospective cohort | 223 biopsy-proven primary IgA nephropathy patients | Serum levels of TNFR1 and TNFR2 | Both TNFRs levels were significantly higher in patients with eGFR < 60 mL/min/1.73 m2 than in patients with higher eGFR. Both TNFRs were associated with renal function decline, independent of age and uric acid levels. | [153] |
| 2015 | Prospective cohort | 262 patients admitted for a CAG and/or a PCI | Serum levels of TNFR1 and TNFR2 | Markedly elevated concentrations of circulating TNFRs were correlated with the occurrence of contrast-induced nephropathy (CIN) and significantly associated with prolonged renal dysfunction, regardless of the development of CIN. | [116] |
| 2015 | Prospective cohort | 131 patients with CKD at stages 4 and 5 | Serum levels of TNFR1 and TNFR2 | Both TNFRs were independently associated with all-cause mortality or an increased risk for cardiovascular events in advanced CKD, irrespective of the cause of kidney disease. | [129] |
| 2015 | Prospective cohort | 347 patients with newly diagnosed biopsy-proven primary IgA nephropt2athy | Plasma levels of TNFR1 and TNFR2 | eGFR decreased and proteinuria worsened proportionally as TNFR1 and TNFR2 levels increased. Tubulointerstitial lesions, such as interstitial fibrosis and tubular atrophy, were significantly more severe as concentrations of circulating TNFRs increased, regardless of eGFR levels. | [154] |
| 2015 | Prospective cohort | 193 Pima Indians with T2D | Serum levels of TNFR1 and TNFR2 | Elevated serum concentrations of TNFR1 or TNFR2 were associated with increased risk of ESRD in American Indians with type 2 diabetes, after accounting for traditional risk factors including UACR and mGFR. | [155] |
| 2015 | Cross- sectional | 106 biopsy-proven IgA nephropathy patients and 34 healthy subjects | Serum and urinary levels of TNFR1 and TNFR2 | Elevated serum TNFR1 or TNFR2 levels were significantly associated with the severity of renal interstitial fibrosis after adjusting for eGFR, UPCR, and other markers of tubular damage. | [112] |
| 2015 | Prospective cohort | 207 patients undergoing HD | Serum levels of TNFR1 and TNFR2 | Prevalent hemodialysis patients had several-fold higher levels of sTNFRs compared to previous studies in CKD stage-4 patients. However, no consistent association between TNFR and mortality was observed. | [131] |
| 2016 | Prospective cohort | 2220 Chinese patients aged 50–70 years old with eGFR > 60 mL/min/1.73 m2 | Plasma levels of TNFR2 | Elevated levels of TNFR2 were independently associated with a greater risk of kidney function decline in middle-aged and elderly Chinese. | [156] |
| 2016 | Prospective cohort | 83 Pima Indians with T2D | Serum levels of TNF-α, TNFR1 and TNFR2 | TNFR1 and TNFR2 significantly correlated inversely with the percentage of endothelial cell fenestration and the total filtration surface per glomerulus. Thus, TNFRs may be involved in the pathogenesis of early glomerular lesions in DN. | [124] |
| 2016 | Prospective cohort | 86 patients with CKD stages 2–4 | A panel of biomarkers, including TNF-α | The panel of proteomic inflammatory and mineral and bone disorder biomarkers showed a better performance in detecting early CKD stages, disease progression, and vascular changes, than each single biomarker. | [134] |
| 2016 | Prospective cohort | 3430 participants with eGFR of 20–70 mL/min/1.73 m2 | Plasma levels of TNF-α | Elevated plasma levels of TNF-α and decreased serum albumin were associated with rapid loss of kidney function in patients with CKD. | [10] |
| 2016 | Prospective cohort | 543 patients with stage 5 CKD | Serum levels TNF-α | TNF-α could, independently of other biomarkers, predict all-cause mortality, but not clinical CVD. | [157] |
| 2016 | Prospective cohort | 607 Swedish patients with T2D | Circulating levels of TNFR1 and TNFR2 | Higher levels of both TNFR1 and TNFR2 were associated with prevalent diabetic kidney disease, as well as with worsened kidney function and higher urinary albumin/creatinine ratio. | [127] |
| 2017 | Nested case-control | 380 participants with early DKD (190 matched case-control pairs) from the ACCORD study | Plasma levels of TNFR1 and TNFR2 | At baseline, median levels of TNFR1 and TNFR2 were roughly two-fold higher in the advanced than in the early cohort. TNFR1 and TNFR2 levels were associated with higher risk of eGFR decline in T2DM persons with both early (ACCORD) and established (VA-NEPHRON-D) DKD. In both cohorts, patients who reached the renal outcome had higher baseline TNFRs levels. | [123] |
| Prospective cohort | 1256 participants with advanced DKD from the VA-NEPHRON-D Cohort | ||||
| 2017 | Prospective cohort | 984 patients with CKD | Serum levels of TNFR1 and TNFR2 | TNFR1 and TNFR2 predicted CVD risk, even after adjustment for clinical covariates, such as urinary protein/creatinine ratio, eGFR, and high-sensitivity CRP. | [113] |
| 2017 | Prospective cohort | 1.135 French patients with T2D | Serum levels of TNFR1 | In addition to established risk factors, TNFR1 improves risk prediction of loss of renal function in patients with T2D. | [125] |
| 2017 | Prospective cohort | 319 patients receiving maintenance hemodialysis | Serum levels of TNF-α, TNFR1 and TNFR2 | Elevated TNFRs levels were associated with an increased risk of cardiovascular and/or all-cause mortality, independently of other studied covariates, in patients undergoing HD. | [132] |
| 2017 | Prospective cohort | 122 patients with confirmed DN | Renal tissue expression of TNFR1 and TNFR2 | No correlations were found between glomerular or tubular expressions of TNFRs, and clinical parameters, including GFR decline slopes. | [158] |
| 2018 | Prospective cohort | 453 Indigenous Australians with and without diabetes and/or CKD | Serum levels of TNFR1 | Circulating levels of TNFR1 were associated with greater kidney disease progression, independently of albuminuria and eGFR, in Indigenous Australians with diabetes. | [126] |
| 2018 | Prospective cohort | 594 Japanese patients with T2D and eGFR > 30 mL/min/1.73 m2 (stages 1 to 3) | Serum levels of TNF-α, TNFR1 and TNFR2 | Circulating TNF-related inflammatory biomarkers were associated with urinary albumin/creatinine ratio and eGFR. Among the biomarkers, the association of TNFRs with eGFR was the strongest after adjustment for relevant covariates. | [111] |
| 2018 | Prospective cohort | 2399 patients with CKD and no history of cardiovascular disease | Plasma levels of TNF-α | A composite inflammation score with 4 biomarkers (IL-6, TNF-a, fibrinogen, and albumin) was associated with a graded increase in risk for incident atherosclerotic vascular disease events and death in patients with CKD. | [23] |
| 2018 | Prospective cohort | 2871 participants multiethnic cohort | Serum levels of TNFR1 | Elevated serum TNFR1 concentrations were associated with faster declines in eGFR over the course of a decade in a multiethnic population, independently of previously known risk factors for kidney disease progression. | [159] |
| 2019 | Prospective cohort | 525 diabetic participants of 3 independent cohorts | 194 proteins, including TNFR1 and TNFR2 | Kidney risk inflammatory signature (KRIS) comprising 17 circulating inflammatory proteins, including TNFR1 and TNFR2, were associated with incident ESRD in diabetic patients. | [136] |
| 2019 | Systematic review and Meta-analysis | 6526 participants from 11 cohorts for TNFR1 measurements and 5385 participants from 10 prospective for TNFR2 measurements | Circulating levels of TNFR1 and TNFR2 | Circulating TNFR-1 and TNFR-2 are reliable predictors of DKD progression. | [160] |
| 2019 | Prospective cohort | 47 patients with diabetes and eGFR > 60 mL/min/1.73 | Serum levels of TNFR1 | In patients with an early decline in renal function, TNFR1 values increased as eGFR decreased, over an 8-year period. In contrast, there were no significant changes in soluble TNFR1 levels in patients with stable renal function. | [161] |
| 2020 | Prospective cohort | 165 case participants from the ADVANCE trial and 330 matched control | Plasma levels of TNFR1 and TNFR2 | Elevated circulating TNFR1 and TNFR2 levels were associated with poor kidney outcome. | [162] |
| 2020 | Cross-sectional | 26 adults with terminal stage CKD and 10 healthy controls | Serum levels of 27 cytokines, including TNF-α | Serum levels of TNF-α were increased 6 to 12 times in patients with CKD, as compared to controls. TNF-α levels positively correlated with complement systems components. | [135] |
| 2020 | Prospective cohort | 894 CRIC Study participants with diabetes and an eGFR of < 60 mL/min/1.73 m2 | Plasma levels of TNFR1 and TNFR2 | Higher plasma levels of TNFR1 and TNFR2 were associated with increased risk of progression of DN. TNFR2 had the highest risk after accounting for the other biomarkers. | [163] |
| 2020 | Prospective cohort | 651 children with 1–16 years old with an eGFR of 30–90 mL/min/1.73 m2 | Plasma levels of TNFR1 and TNFR2 | Children with a plasma TNFR1 or TNFR2 concentration in the highest quartile were at significantly higher risk of CKD progression, compared with children with a concentration for the respective biomarker in the lowest quartile. | [164] |
| 2021 | Prospective cohort | 139 adults with CKD stages 1 to 5 | Serum levels of 11 markers, including TNFR1 and TNFR2 | Patients with high TNFR1, coupled with low complement 3a desarginine, almost universally (96%) developed the composite renal and mortality endpoint. | [165] |
| 2021 | Prospective cohort | 346 T1D patients, 198 with macroalbuminuria and 148 with microalbuminuria | 25 TNF family proteins, including TNFR1 and TNFR2 | Levels of TNR1 and TNFR2 were associated with increased risk of early progressive renal decline in T1D diabetic patients with macro and microalbuminuria. | [137] |
| 2021 | Prospective cohort | 523 CKD patients undergoing kidney biopsy with a diverse set of kidney diseases | Plasma levels of TNFR1 and TNFR2 | Both TNFR1 and TNFR2 were associated with tubulointerstitial and glomerular lesions; each doubling of TNFR1 and TNFR2 was associated with an increased risk of CKD progression, but only TNFR2 was associated with risk of death. | [114] |
| 2021 | Prospective cohort | 2553 patients with T2D and normoalbuminuria | Plasma levels of TNFR1 and TNFR2 | Each doubling of baseline TNFR1 and TNFR2 was associated with a higher risk of kidney outcome (40% reduction in eGFR or kidney failure), in normoalbuminuric patients. | [166] |
| 2021 | Prospective cohort | 3523 participants from the CANVAS placebo-controlled trial | Plasma levels of TNFR1 and TNFR2 | Each doubling in baseline TNFR1 and TNFR2 was associated with a higher risk of kidney outcomes. Early decreases in TNFR1 and TNFR2 during treatment were associated with a lower risk of disease progression. | [167] |
| 2021 | Cross-sectional | 499 patients with T2D and eGFR ≥ 60 mL/min/1.73 m2 | Serum and urinary TNFR1 and TNFR2 levels | Kidney measures appear to be strongly associated with serum TNFRs, rather than urinary TNFRs in patients with type 2 diabetes and normal renal function. | [168] |
| 2021 | Prospective cohort | 594 participants with T2D and eGFR < 60 mL/min/1.73 m2 | Plasma levels of TNFR1 and TNFR2 | TNFR1 and TNFR2 were associated with risk of incident kidney failure needing RRT, in adults with diabetes and an eGFR < 60 mL/min/1.73 m2, after adjustment for established risk factors. | [169] |
| 2021 | Cross-sectional | 5 human renal biopsy specimens from IgA nephropathy patients and 1 healthy control | Transcriptomic analysis of single-cell RNA | Tubular cells of IgA nephropathy patients were enriched in inflammatory pathways, including TNF-α signaling. | [138] |
| 2021 | Prospective cohort | 289 ESRD patients under chronic HD therapy | Several biomarkers circulating levels, including TNFR2 | TNFR2 levels were an independent predictor of all-cause mortality (1-year follow-up study). Circulating levels of cfDNA emerged as the best predictor of mortality. | [133] |
| 2022 | Prospective cohort | 1325 participants from the CANVAS trial with prevalent DKD | KidneyIntelX score, including plasma levels of TNFR1 and TNFR2 | Changes in the KidneyIntelX score from baseline to 1 year were associated with future risk of CKD progression, independently of the baseline risk score and treatment arm. | [128] |
5. Considerations for Future Research
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crews, D.C.; Bello, A.K.; Saadi, G.; World Kidney Day Steering Committee. Burden, access, and disparities in kidney disease. Braz. J. Med. Biol. Res. 2019, 52, e8338. [Google Scholar] [CrossRef] [PubMed]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication—Worldwide more than 850 million individuals have kidney diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.; Stevens, P.E.; Bilous, R.W.; Coresh, J.; De Francisco, A.L.M.; De Jong, P.E.; Griffith, K.E.; Hemmelgarn, B.R.; Iseki, K.; Lamb, E.J.; et al. Kidney disease: Improving global outcomes (KDIGO) CKD work group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- Schlondorff, D.O. Overview of factors contributing to the pathophysiology of progressive renal disease. Kidney Int. 2008, 74, 860–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Zhang, W.R.; Parikh, C.R. Biomarkers of Acute and Chronic Kidney Disease. Annu. Rev. Physiol. 2019, 81, 309–333. [Google Scholar] [CrossRef]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef] [Green Version]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Schei, J.; Stefansson, V.T.; Eriksen, B.O.; Jenssen, T.G.; Solbu, M.D.; Wilsgaard, T.; Melsom, T. Association of TNF Receptor 2 and CRP with GFR Decline in the General Nondiabetic Population. Clin. J. Am. Soc. Nephrol. 2017, 12, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, C.; Noreddin, A.; Nunes, A. Inflammation in Nonimmune-Mediated Chronic Kidney Disease. In Chronic Kidney Disease—From Pathophysiology to Clinical Improvements; Rath, T., Ed.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.-M.; Rusu, E.; Zilisteanu, D.; Necula, L.G.; Anton, G.; Tanase, C. Inflammation-Related Patterns in the Clinical Staging and Severity Assessment of Chronic Kidney Disease. Dis. Markers 2019, 2019, 1814304. [Google Scholar] [CrossRef] [Green Version]
- Valente, M.J.; Rocha, S.; Coimbra, S.; Catarino, C.; Rocha-Pereira, P.; Bronze-da-Rocha, E.; Oliveira, J.G.; Madureira, J.; Fernandes, J.C.; do Sameiro-Faria, M.; et al. Long Pentraxin 3 as a Broader Biomarker for Multiple Risk Factors in End-Stage Renal Disease: Association with All-Cause Mortality. Mediat. Inflamm. 2019, 2019, 3295725. [Google Scholar] [CrossRef] [Green Version]
- Imig, J.D.; Ryan, M.J. Immune and inflammatory role in renal disease. Compr. Physiol. 2013, 3, 957–976. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Raj, D.S.; Pecoits-Filho, R.; Kimmel, P.L. Chapter 17—Inflammation in Chronic Kidney Disease. In Chronic Renal Disease; Kimmel, P.L., Rosenberg, M.E., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 199–212. [Google Scholar] [CrossRef]
- Yao, Q.; Axelsson, J.; Stenvinkel, P.; Lindholm, B. Chronic Systemic Inflammation in Dialysis Patients: An Update on Causes and Consequences. ASAIO J. 2004, 50, Iii–Ivii. [Google Scholar] [CrossRef]
- Avesani, C.M.; Carrero, J.J.; Axelsson, J.; Qureshi, A.R.; Lindholm, B.; Stenvinkel, P. Inflammation and wasting in chronic kidney disease: Partners in crime. Kidney Int. 2006, 70, S8–S13. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, S.; De Martini, N.; Rotondi, S.; Tartaglione, L.; Ureña-Torres, P.; Bover, J.; Pasquali, M. Bone, inflammation and chronic kidney disease. Clin. Chim. Acta 2020, 506, 236–240. [Google Scholar] [CrossRef]
- Amdur, R.L.; Feldman, H.I.; Dominic, E.A.; Anderson, A.H.; Beddhu, S.; Rahman, M.; Wolf, M.; Reilly, M.; Ojo, A.; Townsend, R.R.; et al. Use of Measures of Inflammation and Kidney Function for Prediction of Atherosclerotic Vascular Disease Events and Death in Patients With CKD: Findings From the CRIC Study. Am. J. Kidney Dis. 2019, 73, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, W.; Booz, G.W.; Wang, Y.; Fan, F.; Roman, R.J. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur. J. Pharmacol. 2018, 820, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M. Inflammatory Mediators and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef]
- Hodgkins, K.S.; Schnaper, H.W. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr. Nephrol. 2012, 27, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse origins of the myofibroblast—Implications for kidney fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Sellati, T.J.; Sahay, B. Cells of Innate Immunity: Mechanisms of Activation. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 258–274. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Imaizumi, T.; Itaya, H.; Fujita, K.; Kudoh, D.; Kudoh, S.; Mori, K.; Fujimoto, K.; Matsumiya, T.; Yoshida, H.; Satoh, K. Expression of tumor necrosis factor-alpha in cultured human endothelial cells stimulated with lipopolysaccharide or interleukin-1alpha. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 410–415. [Google Scholar] [CrossRef] [Green Version]
- Fahey, T.J., 3rd; Turbeville, T.; McIntyre, K. Differential TNF secretion by wound fibroblasts compared to normal fibroblasts in response to LPS. J. Surg. Res. 1995, 58, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; McTiernan, C.F.; Frye, C.S.; Demetris, A.J.; Feldman, A.M. Cardiac-specific overexpression of tumor necrosis factor-alpha causes lethal myocarditis in transgenic mice. J. Card. Fail. 1997, 3, 117–124. [Google Scholar] [CrossRef]
- Gahring, L.C.; Carlson, N.G.; Kulmar, R.A.; Rogers, S.W. Neuronal expression of tumor necrosis factor alpha in the murine brain. Neuroimmunomodulation 1996, 3, 289–303. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Göbel, K.; Ruck, T.; Meuth, S.G. Cytokine signaling in multiple sclerosis: Lost in translation. Mult. Scler. 2018, 24, 432–439. [Google Scholar] [CrossRef]
- Schulte, W.; Bernhagen, J.; Bucala, R. Cytokines in sepsis: Potent immunoregulators and potential therapeutic targets—An updated view. Mediat. Inflamm. 2013, 2013, 165974. [Google Scholar] [CrossRef]
- Tinti, F.; Lai, S.; Noce, A.; Rotondi, S.; Marrone, G.; Mazzaferro, S.; Di Daniele, N.; Mitterhofer, A.P. Chronic Kidney Disease as a Systemic Inflammatory Syndrome: Update on Mechanisms Involved and Potential Treatment. Life 2021, 11, 419. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 23. [Google Scholar] [CrossRef]
- Medler, J.; Wajant, H. Tumor necrosis factor receptor-2 (TNFR2): An overview of an emerging drug target. Expert Opin. Ther. Targets 2019, 23, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Pennica, D.; Goeddel, D.V. Ligand passing: The 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J. Biol. Chem. 1993, 268, 18542–18548. [Google Scholar] [CrossRef]
- Aderka, D.; Engelmann, H.; Maor, Y.; Brakebusch, C.; Wallach, D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J. Exp. Med. 1992, 175, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xanthoulea, S.; Pasparakis, M.; Kousteni, S.; Brakebusch, C.; Wallach, D.; Bauer, J.; Lassmann, H.; Kollias, G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 2004, 200, 367–376. [Google Scholar] [CrossRef]
- Dembic, Z.; Loetscher, H.; Gubler, U.; Pan, Y.C.; Lahm, H.W.; Gentz, R.; Brockhaus, M.; Lesslauer, W. Two human TNF receptors have similar extracellular, but distinct intracellular, domain sequences. Cytokine 1990, 2, 231–237. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.; Huang, J.; Shu, H.B.; Baichwal, V.; Goeddel, D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.; Shu, H.B.; Pan, M.G.; Goeddel, D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996, 84, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, P.N. RIP kinases: Key decision makers in cell death and innate immunity. Cell Death Differ. 2015, 22, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, D.J.; Cheung, H.H.; Mrad, R.L.; Plenchette, S.; Simard, C.; Enwere, E.; Arora, V.; Mak, T.W.; Lacasse, E.C.; Waring, J.; et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2008, 105, 11778–11783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Scheurich, P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J. 2011, 278, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.T.; Hsu, P.-H.; Hsu, W.-C.; Chen, N.-J.; Tseng, P.-H. Polyubiquitination of Transforming Growth Factor β-activated Kinase 1 (TAK1) at Lysine 562 Residue Regulates TLR4-mediated JNK and p38 MAPK Activation. Sci. Rep. 2015, 5, 12300. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Srinivasula, S.M. Ubiquitination and TNFR1 signaling. Results Probl. Cell Differ. 2009, 49, 87–114. [Google Scholar] [CrossRef]
- Li, X.; Zhang, M.; Huang, X.; Liang, W.; Li, G.; Lu, X.; Li, Y.; Pan, H.; Shi, L.; Zhu, H.; et al. Ubiquitination of RIPK1 regulates its activation mediated by TNFR1 and TLRs signaling in distinct manners. Nat. Commun. 2020, 11, 6364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Webster, J.D.; Dugger, D.L.; Goncharov, T.; Roose-Girma, M.; Hung, J.; Kwon, Y.C.; Vucic, D.; Newton, K.; Dixit, V.M. Ubiquitin Ligases cIAP1 and cIAP2 Limit Cell Death to Prevent Inflammation. Cell Rep. 2019, 27, 2679–2689.e2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, K. RIPK1 and RIPK3: Critical regulators of inflammation and cell death. Trends Cell Biol. 2015, 25, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tong, A.; Zhang, Q.; Wei, Y.; Wei, X. The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. J. Mol. Cell Biol. 2020, 13, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Rothe, M.; Pan, M.G.; Henzel, W.J.; Ayres, T.M.; Goeddel, D.V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995, 83, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Borghi, A.; Haegman, M.; Fischer, R.; Carpentier, I.; Bertrand, M.J.M.; Libert, C.; Afonina, I.S.; Beyaert, R. The E3 ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2 signaling complex and mediate TNFR2-induced canonical NF-κB signaling. Biochem. Pharmacol. 2018, 153, 292–298. [Google Scholar] [CrossRef]
- Rauert, H.; Wicovsky, A.; Müller, N.; Siegmund, D.; Spindler, V.; Waschke, J.; Kneitz, C.; Wajant, H. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 2010, 285, 7394–7404. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Thommesen, L.; Laegreid, A. Distinct differences between TNF receptor 1- and TNF receptor 2-mediated activation of NFkappaB. J. Biochem. Mol. Biol. 2005, 38, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Al-Lamki, R.S.; Wang, J.; Vandenabeele, P.; Bradley, J.A.; Thiru, S.; Luo, D.; Min, W.; Pober, J.S.; Bradley, J.R. TNFR1- and TNFR2-mediated signaling pathways in human kidney are cell type-specific and differentially contribute to renal injury. FASEB J. 2005, 19, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, C.; Shamoon, B.; Shyamala, V.; Williams, L.T. Tumor necrosis factor alpha-induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO J. 1997, 16, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.; Li, Y.; Wan, T.; Wang, J.; Zhang, H.; Chen, H.; Min, W. Both internalization and AIP1 association are required for tumor necrosis factor receptor 2-mediated JNK signaling. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, T.; Croft, M. Regulation of PI-3-Kinase and Akt Signaling in T Lymphocytes and Other Cells by TNFR Family Molecules. Front. Immunol. 2013, 4, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, D.; Ernandez, T.; Rosetti, F.; Batal, I.; Cullere, X.; Luscinskas, F.W.; Zhang, Y.; Stavrakis, G.; García-Cardeña, G.; Horwitz, B.H.; et al. Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-β autocrine signaling to promote monocyte recruitment. Immunity 2013, 38, 1025–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fotin-Mleczek, M.; Henkler, F.; Samel, D.; Reichwein, M.; Hausser, A.; Parmryd, I.; Scheurich, P.; Schmid, J.A.; Wajant, H. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J. Cell Sci. 2002, 115, 2757–2770. [Google Scholar] [CrossRef]
- Pimentel-Muiños, F.X.; Seed, B. Regulated commitment of TNF receptor signaling: A molecular switch for death or activation. Immunity 1999, 11, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Naudé, P.J.; den Boer, J.A.; Luiten, P.G.; Eisel, U.L. Tumor necrosis factor receptor cross-talk. FEBS J. 2011, 278, 888–898. [Google Scholar] [CrossRef]
- Tomosugi, N.I.; Cashman, S.J.; Hay, H.; Pusey, C.D.; Evans, D.J.; Shaw, A.; Rees, A.J. Modulation of antibody-mediated glomerular injury in vivo by bacterial lipopolysaccharide, tumor necrosis factor, and IL-1. J. Immunol. 1989, 142, 3083–3090. [Google Scholar]
- Al-Lamki, R.S.; Mayadas, T.N. TNF receptors: Signaling pathways and contribution to renal dysfunction. Kidney Int. 2015, 87, 281–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Lamki, R.S.; Wang, J.; Skepper, J.N.; Thiru, S.; Pober, J.S.; Bradley, J.R. Expression of tumor necrosis factor receptors in normal kidney and rejecting renal transplants. Lab. Investig. 2001, 81, 1503–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, S.; Garrido, P.; Fernandes, J.; Vala, H.; Rocha-Pereira, P.; Costa, E.; Belo, L.; Reis, F.; Santos-Silva, A. Renal risk-benefit determinants of recombinant human erythropoietin therapy in the remnant kidney rat model—Hypertension, anaemia, inflammation and drug dose. Clin. Exp. Pharmacol. Physiol. 2016, 43, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, M.; Haas, C.; Marino, M.; Ryffel, B. Prevention of crescentic glomerulonephritis induced by anti-glomerular membrane antibody in tumor necrosis factor-deficient mice. Lab. Investig. 1998, 78, 1625–1631. [Google Scholar]
- Karkar, A.M.; Smith, J.; Pusey, C.D. Prevention and treatment of experimental crescentic glomerulonephritis by blocking tumour necrosis factor-alpha. Nephrol. Dial. Transplant. 2001, 16, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Misseri, R.; Meldrum, D.R.; Dinarello, C.A.; Dagher, P.; Hile, K.L.; Rink, R.C.; Meldrum, K.K. TNF-alpha mediates obstruction-induced renal tubular cell apoptosis and proapoptotic signaling. Am. J. Physiol.-Ren. Physiol. 2005, 288, F406–F411. [Google Scholar] [CrossRef] [Green Version]
- Meldrum, K.K.; Misseri, R.; Metcalfe, P.; Dinarello, C.A.; Hile, K.L.; Meldrum, D.R. TNF-alpha neutralization ameliorates obstruction-induced renal fibrosis and dysfunction. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 292, R1456–R1464. [Google Scholar] [CrossRef]
- Chen, C.C.; Pedraza, P.L.; Hao, S.; Stier, C.T.; Ferreri, N.R. TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am. J. Physiol.-Ren. Physiol. 2010, 299, F1141–F1150. [Google Scholar] [CrossRef] [Green Version]
- Vielhauer, V.; Stavrakis, G.; Mayadas, T.N. Renal cell-expressed TNF receptor 2, not receptor 1, is essential for the development of glomerulonephritis. J. Clin. Investig. 2005, 115, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Morrissey, J.; McCracken, R.; Tolley, T.; Klahr, S. Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am. J. Physiol. 1999, 277, F766–F772. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am. J. Physiol.-Ren. Physiol. 2003, 285, F610–F618. [Google Scholar] [CrossRef]
- Mulay, S.R.; Eberhard, J.N.; Desai, J.; Marschner, J.A.; Kumar, S.V.; Weidenbusch, M.; Grigorescu, M.; Lech, M.; Eltrich, N.; Müller, L.; et al. Hyperoxaluria Requires TNF Receptors to Initiate Crystal Adhesion and Kidney Stone Disease. J. Am. Soc. Nephrol. 2017, 28, 761–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinkhammer, B.M.; Djudjaj, S.; Kunter, U.; Palsson, R.; Edvardsson, V.O.; Wiech, T.; Thorsteinsdottir, M.; Hardarson, S.; Foresto-Neto, O.; Mulay, S.R.; et al. Cellular and Molecular Mechanisms of Kidney Injury in 2,8-Dihydroxyadenine Nephropathy. J. Am. Soc. Nephrol. 2020, 31, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Liang, R.; Huang, B.; Hou, J.; Yin, J.; Zhao, T.; Zhou, L.; Wu, R.; Qian, Y.; Wang, F. Tumor necrosis factor-α blockade ameliorates diabetic nephropathy in rats. Clin. Kidney J. 2019, 14, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiPetrillo, K.; Coutermarsh, B.; Gesek, F.A. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am. J. Physiol.-Ren. Physiol. 2003, 284, F113–F121. [Google Scholar] [CrossRef] [Green Version]
- Omote, K.; Gohda, T.; Murakoshi, M.; Sasaki, Y.; Kazuno, S.; Fujimura, T.; Ishizaka, M.; Sonoda, Y.; Tomino, Y. Role of the TNF pathway in the progression of diabetic nephropathy in KK-A(y) mice. Am. J. Physiol.-Ren. Physiol. 2014, 306, F1335–F1347. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, Y.; Inokuchi, T.; Yamamoto, A.; Ka, T.; Tsutsumi, Z.; Takahashi, S.; Yamamoto, T. Effect of TNF-alpha inhibition on urinary albumin excretion in experimental diabetic rats. Acta Diabetol. 2007, 44, 215–218. [Google Scholar] [CrossRef]
- Granata, S.; Dalla Gassa, A.; Bellin, G.; Lupo, A.; Zaza, G. Transcriptomics: A Step behind the Comprehension of the Polygenic Influence on Oxidative Stress, Immune Deregulation, and Mitochondrial Dysfunction in Chronic Kidney Disease. Bio. Res. Int. 2016, 2016, 9290857. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, L.M.S.; Liu, J.; Koppitch, K.; Cippà, P.E.; McMahon, A.P. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc. Natl. Acad. Sci. USA 2021, 118, e2026684118. [Google Scholar] [CrossRef]
- Khan, S.B.; Cook, H.T.; Bhangal, G.; Smith, J.; Tam, F.W.; Pusey, C.D. Antibody blockade of TNF-alpha reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int. 2005, 67, 1812–1820. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, U.; Bergler, T.; Rihm, M.; Pace, C.; Krüger, B.; Rümmele, P.; Stoelcker, B.; Banas, B.; Männel, D.N.; Krämer, B.K. Upregulation of TNF receptor type 2 in human and experimental renal allograft rejection. Am. J. Transplant. 2009, 9, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Gai, Z.; Tanishima, H.; Kawakatsu, M.; Itoh, S.; Hatamura, I.; Muragaki, Y. TNF-alpha deficiency accelerates renal tubular interstitial fibrosis in the late stage of ureteral obstruction. Exp. Mol. Pathol. 2008, 85, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Jacob, N.; Yang, H.; Pricop, L.; Liu, Y.; Gao, X.; Zheng, S.G.; Wang, J.; Gao, H.X.; Putterman, C.; Koss, M.N.; et al. Accelerated pathological and clinical nephritis in systemic lupus erythematosus-prone New Zealand Mixed 2328 mice doubly deficient in TNF receptor 1 and TNF receptor 2 via a Th17-associated pathway. J. Immunol. 2009, 182, 2532–2541. [Google Scholar] [CrossRef]
- Taubitz, A.; Schwarz, M.; Eltrich, N.; Lindenmeyer, M.T.; Vielhauer, V. Distinct contributions of TNF receptor 1 and 2 to TNF-induced glomerular inflammation in mice. PLoS ONE 2013, 8, e68167. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Chang, A.; Hack, B.K.; Eadon, M.T.; Alper, S.L.; Cunningham, P.N. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014, 85, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halwachs, G.; Tiran, A.; Reisinger, E.C.; Zach, R.; Sabin, K.; Fölsch, B.; Lanzer, H.; Holzer, H.; Wilders-Truschnig, M. Serum levels of the soluble receptor for tumor necrosis factor in patients with renal disease. Clin. Investig. 1994, 72, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Jhangri, G.S.; Curhan, G. Biomarkers of inflammation and progression of chronic kidney disease. Kidney Int. 2005, 68, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Kamei, N.; Yamashita, M.; Nishizaki, Y.; Yanagisawa, N.; Nojiri, S.; Tanaka, K.; Yamashita, Y.; Shibata, T.; Murakoshi, M.; Suzuki, Y.; et al. Association between circulating tumor necrosis factor-related biomarkers and estimated glomerular filtration rate in type 2 diabetes. Sci. Rep. 2018, 8, 15302. [Google Scholar] [CrossRef]
- Sonoda, Y.; Gohda, T.; Suzuki, Y.; Omote, K.; Ishizaka, M.; Matsuoka, J.; Tomino, Y. Circulating TNF receptors 1 and 2 are associated with the severity of renal interstitial fibrosis in IgA nephropathy. PLoS ONE 2015, 10, e0122212. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.; Cha, R.H.; Kim, Y.C.; An, J.N.; Kim, D.K.; Yoo, K.D.; Lee, S.M.; Kim, M.H.; Park, J.T.; Kang, S.W.; et al. Circulating TNF receptors predict cardiovascular disease in patients with chronic kidney disease. Medicine 2017, 96, e6666. [Google Scholar] [CrossRef]
- Srivastava, A.; Schmidt, I.M.; Palsson, R.; Weins, A.; Bonventre, J.V.; Sabbisetti, V.; Stillman, I.E.; Rennke, H.G.; Waikar, S.S. The Associations of Plasma Biomarkers of Inflammation With Histopathologic Lesions, Kidney Disease Progression, and Mortality-The Boston Kidney Biopsy Cohort Study. Kidney Int. Rep. 2021, 6, 685–694. [Google Scholar] [CrossRef]
- Lee, S.M.; Yang, S.; Cha, R.H.; Kim, M.; An, J.N.; Paik, J.H.; Kim, D.K.; Kang, S.W.; Lim, C.S.; Kim, Y.S.; et al. Circulating TNF receptors are significant prognostic biomarkers for idiopathic membranous nephropathy. PLoS ONE 2014, 9, e104354. [Google Scholar] [CrossRef]
- An, J.N.; Yoo, K.D.; Hwang, J.H.; Kim, H.L.; Kim, S.H.; Yang, S.H.; Kim, J.H.; Kim, D.K.; Oh, Y.K.; Kim, Y.S.; et al. Circulating tumour necrosis factor receptors 1 and 2 predict contrast-induced nephropathy and progressive renal dysfunction: A prospective cohort study. Nephrology 2015, 20, 552–559. [Google Scholar] [CrossRef]
- Wu, T.; Xie, C.; Wang, H.W.; Zhou, X.J.; Schwartz, N.; Calixto, S.; Mackay, M.; Aranow, C.; Putterman, C.; Mohan, C. Elevated urinary VCAM-1, P-selectin, soluble TNF receptor-1, and CXC chemokine ligand 16 in multiple murine lupus strains and human lupus nephritis. J. Immunol. 2007, 179, 7166–7175. [Google Scholar] [CrossRef] [Green Version]
- Koenig, K.F.; Groeschl, I.; Pesickova, S.S.; Tesar, V.; Eisenberger, U.; Trendelenburg, M. Serum cytokine profile in patients with active lupus nephritis. Cytokine 2012, 60, 410–416. [Google Scholar] [CrossRef]
- Murakoshi, M.; Gohda, T.; Suzuki, Y. Circulating Tumor Necrosis Factor Receptors: A Potential Biomarker for the Progression of Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 1957. [Google Scholar] [CrossRef] [Green Version]
- Gohda, T.; Niewczas, M.A.; Ficociello, L.H.; Walker, W.H.; Skupien, J.; Rosetti, F.; Cullere, X.; Johnson, A.C.; Crabtree, G.; Smiles, A.M.; et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J. Am. Soc. Nephrol. 2012, 23, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef]
- Krolewski, A.S.; Niewczas, M.A.; Skupien, J.; Gohda, T.; Smiles, A.; Eckfeldt, J.H.; Doria, A.; Warram, J.H. Early progressive renal decline precedes the onset of microalbuminuria and its progression to macroalbuminuria. Diabetes Care 2014, 37, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Coca, S.G.; Nadkarni, G.N.; Huang, Y.; Moledina, D.G.; Rao, V.; Zhang, J.; Ferket, B.; Crowley, S.T.; Fried, L.F.; Parikh, C.R. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2786–2793. [Google Scholar] [CrossRef] [Green Version]
- Pavkov, M.E.; Weil, E.J.; Fufaa, G.D.; Nelson, R.G.; Lemley, K.V.; Knowler, W.C.; Niewczas, M.A.; Krolewski, A.S. Tumor necrosis factor receptors 1 and 2 are associated with early glomerular lesions in type 2 diabetes. Kidney Int. 2016, 89, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Saulnier, P.J.; Gand, E.; Velho, G.; Mohammedi, K.; Zaoui, P.; Fraty, M.; Halimi, J.M.; Roussel, R.; Ragot, S.; Hadjadj, S. Association of Circulating Biomarkers (Adrenomedullin, TNFR1, and NT-proBNP) With Renal Function Decline in Patients With Type 2 Diabetes: A French Prospective Cohort. Diabetes Care 2017, 40, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Barr, E.L.M.; Barzi, F.; Hughes, J.T.; Jerums, G.; Hoy, W.E.; O’Dea, K.; Jones, G.R.D.; Lawton, P.D.; Brown, A.D.H.; Thomas, M.; et al. High Baseline Levels of Tumor Necrosis Factor Receptor 1 Are Associated With Progression of Kidney Disease in Indigenous Australians With Diabetes: The eGFR Follow-up Study. Diabetes Care 2018, 41, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, A.C.; Östgren, C.J.; Nystrom, F.H.; Länne, T.; Jennersjö, P.; Larsson, A.; Ärnlöv, J. Association of soluble tumor necrosis factor receptors 1 and 2 with nephropathy, cardiovascular events, and total mortality in type 2 diabetes. Cardiovasc. Diabetol. 2016, 15, 40. [Google Scholar] [CrossRef] [Green Version]
- Lam, D.; Nadkarni, G.N.; Mosoyan, G.; Neal, B.; Mahaffey, K.W.; Rosenthal, N.; Hansen, M.K.; Heerspink, H.J.L.; Fleming, F.; Coca, S.G. Clinical Utility of KidneyIntelX in Early Stages of Diabetic Kidney Disease in the CANVAS Trial. Am. J. Nephrol. 2022, 53, 21–31. [Google Scholar] [CrossRef]
- Neirynck, N.; Glorieux, G.; Schepers, E.; Verbeke, F.; Vanholder, R. Soluble tumor necrosis factor receptor 1 and 2 predict outcomes in advanced chronic kidney disease: A prospective cohort study. PLoS ONE 2015, 10, e0122073. [Google Scholar] [CrossRef] [Green Version]
- Saulnier, P.J.; Gand, E.; Ragot, S.; Ducrocq, G.; Halimi, J.M.; Hulin-Delmotte, C.; Llaty, P.; Montaigne, D.; Rigalleau, V.; Roussel, R.; et al. Association of serum concentration of TNFR1 with all-cause mortality in patients with type 2 diabetes and chronic kidney disease: Follow-up of the SURDIAGENE Cohort. Diabetes Care 2014, 37, 1425–1431. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, A.C.; Carrero, J.J.; Stenvinkel, P.; Bottai, M.; Barany, P.; Larsson, A.; Ärnlöv, J. High levels of soluble tumor necrosis factor receptors 1 and 2 and their association with mortality in patients undergoing hemodialysis. Cardiorenal Med. 2015, 5, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Gohda, T.; Maruyama, S.; Kamei, N.; Yamaguchi, S.; Shibata, T.; Murakoshi, M.; Horikoshi, S.; Tomino, Y.; Ohsawa, I.; Gotoh, H.; et al. Circulating TNF Receptors 1 and 2 Predict Mortality in Patients with End-stage Renal Disease Undergoing Dialysis. Sci. Rep. 2017, 7, 43520. [Google Scholar] [CrossRef]
- Coimbra, S.; Rocha, S.; Nascimento, H.; Valente, M.J.; Catarino, C.; Rocha-Pereira, P.; Sameiro-Faria, M.; Oliveira, J.G.; Madureira, J.; Fernandes, J.C.; et al. Cell-free DNA as a marker for the outcome of end-stage renal disease patients on haemodialysis. Clin. Kidney J. 2021, 14, 1371–1378. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.-M.; Rusu, E.; Zilisteanu, D.; Albulescu, R.; Anton, G.; Tanase, C. Proteomic Biomarkers Panel: New Insights in Chronic Kidney Disease. Dis. Markers 2016, 2016, 3185232. [Google Scholar] [CrossRef] [Green Version]
- Romanova, Y.; Laikov, A.; Markelova, M.; Khadiullina, R.; Makseev, A.; Hasanova, M.; Rizvanov, A.; Khaiboullina, S.; Salafutdinov, I. Proteomic Analysis of Human Serum from Patients with Chronic Kidney Disease. Biomolecules 2020, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Md Dom, Z.I.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.J.; et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Ihara, K.; Skupien, J.; Krolewski, B.; Md Dom, Z.I.; O’Neil, K.; Satake, E.; Kobayashi, H.; Rashidi, N.M.; Niewczas, M.A.; Krolewski, A.S. A profile of multiple circulating tumor necrosis factor receptors associated with early progressive kidney decline in Type 1 Diabetes is similar to profiles in autoimmune disorders. Kidney Int. 2021, 99, 725–736. [Google Scholar] [CrossRef]
- Tang, R.; Meng, T.; Lin, W.; Shen, C.; Ooi, J.D.; Eggenhuizen, P.J.; Jin, P.; Ding, X.; Chen, J.; Tang, Y.; et al. A Partial Picture of the Single-Cell Transcriptomics of Human IgA Nephropathy. Front. Immunol. 2021, 12, 645988. [Google Scholar] [CrossRef]
- Premužić, V.; Padjen, I.; Cerovec, M.; Ćorić, M.; Jelaković, B.; Anić, B. The Association of TNF-Alpha Inhibitors and Development of IgA Nephropathy in Patients with Rheumatoid Arthritis and Diabetes. Case Rep. Nephrol. 2020, 2020, 9480860. [Google Scholar] [CrossRef]
- Stokes, M.B.; Foster, K.; Markowitz, G.S.; Ebrahimi, F.; Hines, W.; Kaufman, D.; Moore, B.; Wolde, D.; D’Agati, V.D. Development of glomerulonephritis during anti-TNF-alpha therapy for rheumatoid arthritis. Nephrol. Dial. Transplant. 2005, 20, 1400–1406. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Lee, C.K.; Cha, H.S.; Choe, J.Y.; Park, E.J.; Kim, J. Effect of anti-tumor necrosis factor alpha treatment of rheumatoid arthritis and chronic kidney disease. Rheumatol. Int. 2015, 35, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Bartolucci, P.; Ramanoelina, J.; Cohen, P.; Mahr, A.; Godmer, P.; Le Hello, C.; Guillevin, L. Efficacy of the anti-TNF-alpha antibody infliximab against refractory systemic vasculitides: An open pilot study on 10 patients. Rheumatology 2002, 41, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Garrouste, C.; Anglicheau, D.; Kamar, N.; Bachelier, C.; Rivalan, J.; Pereira, B.; Caillard, S.; Aniort, J.; Gatault, P.; Soubrier, M.; et al. Anti-TNFα therapy for chronic inflammatory disease in kidney transplant recipients: Clinical outcomes. Medicine 2016, 95, e5108. [Google Scholar] [CrossRef]
- Keller, C.R.; Odden, M.C.; Fried, L.F.; Newman, A.B.; Angleman, S.; Green, C.A.; Cummings, S.R.; Harris, T.B.; Shlipak, M.G. Kidney function and markers of inflammation in elderly persons without chronic kidney disease: The health, aging, and body composition study. Kidney Int. 2007, 71, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Keller, C.; Katz, R.; Cushman, M.; Fried, L.F.; Shlipak, M. Association of kidney function with inflammatory and procoagulant markers in a diverse cohort: A cross-sectional analysis from the Multi-Ethnic Study of Atherosclerosis (MESA). BMC Nephrol. 2008, 9, 9. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Ficociello, L.H.; Johnson, A.C.; Walker, W.; Rosolowsky, E.T.; Roshan, B.; Warram, J.H.; Krolewski, A.S. Serum concentrations of markers of TNFalpha and Fas-mediated pathways and renal function in nonproteinuric patients with type 1 diabetes. Clin. J. Am. Soc. Nephrol. 2009, 4, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Idasiak-Piechocka, I.; Oko, A.; Pawliczak, E.; Kaczmarek, E.; Czekalski, S. Urinary excretion of soluble tumour necrosis factor receptor 1 as a marker of increased risk of progressive kidney function deterioration in patients with primary chronic glomerulonephritis. Nephrol. Dial. Transplant. 2010, 25, 3948–3956. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, A.; Larson, M.G.; Guo, C.Y.; Vasan, R.S.; Lipinska, I.; O’Donnell, C.J.; Kathiresan, S.; Meigs, J.B.; Keaney, J.F., Jr.; Rong, J.; et al. Inflammation, kidney function and albuminuria in the Framingham Offspring cohort. Nephrol. Dial. Transplant. 2011, 26, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Shankar, A.; Sun, L.; Klein, B.E.; Lee, K.E.; Muntner, P.; Nieto, F.J.; Tsai, M.Y.; Cruickshanks, K.J.; Schubert, C.R.; Brazy, P.C.; et al. Markers of inflammation predict the long-term risk of developing chronic kidney disease: A population-based cohort study. Kidney Int. 2011, 80, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Zwiech, R. Predictive value of conjointly examined IL-1ra, TNF-R I, TNF-R II, and RANTES in patients with primary glomerulonephritis. J. Korean Med. Sci. 2013, 28, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Forsblom, C.; Moran, J.; Harjutsalo, V.; Loughman, T.; Wadén, J.; Tolonen, N.; Thorn, L.; Saraheimo, M.; Gordin, D.; Groop, P.H.; et al. Added value of soluble tumor necrosis factor-α receptor 1 as a biomarker of ESRD risk in patients with type 1 diabetes. Diabetes Care 2014, 37, 2334–2342. [Google Scholar] [CrossRef] [Green Version]
- Skupien, J.; Warram, J.H.; Niewczas, M.A.; Gohda, T.; Malecki, M.; Mychaleckyj, J.C.; Galecki, A.T.; Krolewski, A.S. Synergism between circulating tumor necrosis factor receptor 2 and HbA(1c) in determining renal decline during 5-18 years of follow-up in patients with type 1 diabetes and proteinuria. Diabetes Care 2014, 37, 2601–2608. [Google Scholar] [CrossRef] [Green Version]
- Murakoshi, M.; Gohda, T.; Sonoda, Y.; Suzuki, H.; Tomino, Y.; Horikoshi, S.; Suzuki, Y. Effect of tonsillectomy with steroid pulse therapy on circulating tumor necrosis factor receptors 1 and 2 in IgA nephropathy. Clin. Exp. Nephrol. 2017, 21, 1068–1074. [Google Scholar] [CrossRef]
- Oh, Y.J.; An, J.N.; Kim, C.T.; Yang, S.H.; Lee, H.; Kim, D.K.; Joo, K.W.; Paik, J.H.; Kang, S.W.; Park, J.T.; et al. Circulating Tumor Necrosis Factor α Receptors Predict the Outcomes of Human IgA Nephropathy: A Prospective Cohort Study. PLoS ONE 2015, 10, e0132826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavkov, M.E.; Nelson, R.G.; Knowler, W.C.; Cheng, Y.; Krolewski, A.S.; Niewczas, M.A. Elevation of circulating TNF receptors 1 and 2 increases the risk of end-stage renal disease in American Indians with type 2 diabetes. Kidney Int. 2015, 87, 812–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Deng, Y.; Sun, L.; Ye, X.; Yao, P.; Hu, Y.; Wang, F.; Ma, Y.; Li, H.; Liu, Y.; et al. Elevated plasma tumor necrosis factor-α receptor 2 and resistin are associated with increased incidence of kidney function decline in Chinese adults. Endocrine 2016, 52, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Axelsson, J.; Machowska, A.; Heimbürger, O.; Bárány, P.; Lindholm, B.; Lindström, K.; Stenvinkel, P.; Qureshi, A.R. Biomarkers of Cardiovascular Disease and Mortality Risk in Patients with Advanced CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Park, J.; Kim, J.; Jang, H.R.; Kwon, G.Y.; Huh, W.; Kim, Y.G.; Kim, D.J.; Oh, H.Y.; Lee, J.E. Tissue expression of tubular injury markers is associated with renal function decline in diabetic nephropathy. J. Diabetes Complicat. 2017, 31, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Bhatraju, P.K.; Zelnick, L.R.; Shlipak, M.; Katz, R.; Kestenbaum, B. Association of Soluble TNFR-1 Concentrations with Long-Term Decline in Kidney Function: The Multi-Ethnic Study of Atherosclerosis. J. Am. Soc. Nephrol. 2018, 29, 2713–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Luo, T.; Wang, K.; Wang, Y.; Yang, S.; Li, Q.; Hu, J. Circulating TNF receptors 1 and 2 predict progression of diabetic kidney disease: A meta-analysis. Diabetes Metab. Res. Rev. 2019, 35, e3195. [Google Scholar] [CrossRef]
- MacIsaac, R.J.; Farag, M.; Obeyesekere, V.; Clarke, M.; Boston, R.; Ward, G.M.; Jerums, G.; Ekinci, E.I. Changes in soluble tumor necrosis factor receptor type 1 levels and early renal function decline in patients with diabetes. J. Diabetes Investig. 2019, 10, 1537–1542. [Google Scholar] [CrossRef]
- Oshima, M.; Hara, A.; Toyama, T.; Jun, M.; Pollock, C.; Jardine, M.; Harrap, S.; Poulter, N.; Cooper, M.E.; Woodward, M.; et al. Comparison of Circulating Biomarkers in Predicting Diabetic Kidney Disease Progression with Autoantibodies to Erythropoietin Receptor. Kidney Int. Rep. 2021, 6, 284–295. [Google Scholar] [CrossRef]
- Schrauben, S.J.; Shou, H.; Zhang, X.; Anderson, A.H.; Bonventre, J.V.; Chen, J.; Coca, S.; Furth, S.L.; Greenberg, J.H.; Gutierrez, O.M.; et al. Association of Multiple Plasma Biomarker Concentrations with Progression of Prevalent Diabetic Kidney Disease: Findings from the Chronic Renal Insufficiency Cohort (CRIC) Study. J. Am. Soc. Nephrol. 2021, 32, 115–126. [Google Scholar] [CrossRef]
- Greenberg, J.H.; Abraham, A.G.; Xu, Y.; Schelling, J.R.; Feldman, H.I.; Sabbisetti, V.S.; Gonzalez, M.C.; Coca, S.; Schrauben, S.J.; Waikar, S.S.; et al. Plasma Biomarkers of Tubular Injury and Inflammation Are Associated with CKD Progression in Children. J. Am. Soc. Nephrol. 2020, 31, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.P.; Conroy, C.; Naicker, S.D.; Cormican, S.; Griffin, T.P.; Islam, M.N.; McCole, E.M.; McConnell, I.; Lamont, J.; FitzGerald, P.; et al. Multiplex Serum Biomarker Assays Improve Prediction of Renal and Mortality Outcomes in Chronic Kidney Disease. Kidney360 2021, 2, 1225–1239. [Google Scholar] [CrossRef] [PubMed]
- Waijer, S.W.; Sen, T.; Arnott, C.; Neal, B.; Kosterink, J.G.W.; Mahaffey, K.W.; Parikh, C.R.; de Zeeuw, D.; Perkovic, V.; Neuen, B.L.; et al. Association between TNF Receptors and KIM-1 with Kidney Outcomes in Early-Stage Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2021, 17, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Li, J.; Neuen, B.L.; Neal, B.; Arnott, C.; Parikh, C.R.; Coca, S.G.; Perkovic, V.; Mahaffey, K.W.; Yavin, Y.; et al. Effects of the SGLT2 inhibitor canagliflozin on plasma biomarkers TNFR-1, TNFR-2 and KIM-1 in the CANVAS trial. Diabetologia 2021, 64, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Gohda, T.; Kamei, N.; Kubota, M.; Tanaka, K.; Yamashita, Y.; Sakuma, H.; Kishida, C.; Adachi, E.; Koshida, T.; Murakoshi, M.; et al. Fractional excretion of tumor necrosis factor receptor 1 and 2 in patients with type 2 diabetes and normal renal function. J. Diabetes Investig. 2021, 12, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M.; Shlipak, M.G.; Katz, R.; Waikar, S.S.; Greenberg, J.H.; Schrauben, S.J.; Coca, S.; Parikh, C.R.; Vasan, R.S.; Feldman, H.I.; et al. Associations of Plasma Biomarkers of Inflammation, Fibrosis, and Kidney Tubular Injury With Progression of Diabetic Kidney Disease: A Cohort Study. Am. J. Kidney Dis. 2021, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Udalova, I.; Monaco, C.; Nanchahal, J.; Feldmann, M. Anti-TNF Therapy. Microbiol. Spectr. 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lousa, I.; Reis, F.; Santos-Silva, A.; Belo, L. The Signaling Pathway of TNF Receptors: Linking Animal Models of Renal Disease to Human CKD. Int. J. Mol. Sci. 2022, 23, 3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063284
Lousa I, Reis F, Santos-Silva A, Belo L. The Signaling Pathway of TNF Receptors: Linking Animal Models of Renal Disease to Human CKD. International Journal of Molecular Sciences. 2022; 23(6):3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063284
Chicago/Turabian StyleLousa, Irina, Flávio Reis, Alice Santos-Silva, and Luís Belo. 2022. "The Signaling Pathway of TNF Receptors: Linking Animal Models of Renal Disease to Human CKD" International Journal of Molecular Sciences 23, no. 6: 3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063284