
Porcine Circovirus Type 4 Strains Circulating in China Are Relatively Stable and Have Higher Homology with Mink Circovirus than Other Porcine Circovirus Types

 ,
,
Abstract
:1. Introduction
2. Results
2.1. The Genome and Protein Structure of PCV4 Strains Circulating in China Are Relatively Stable
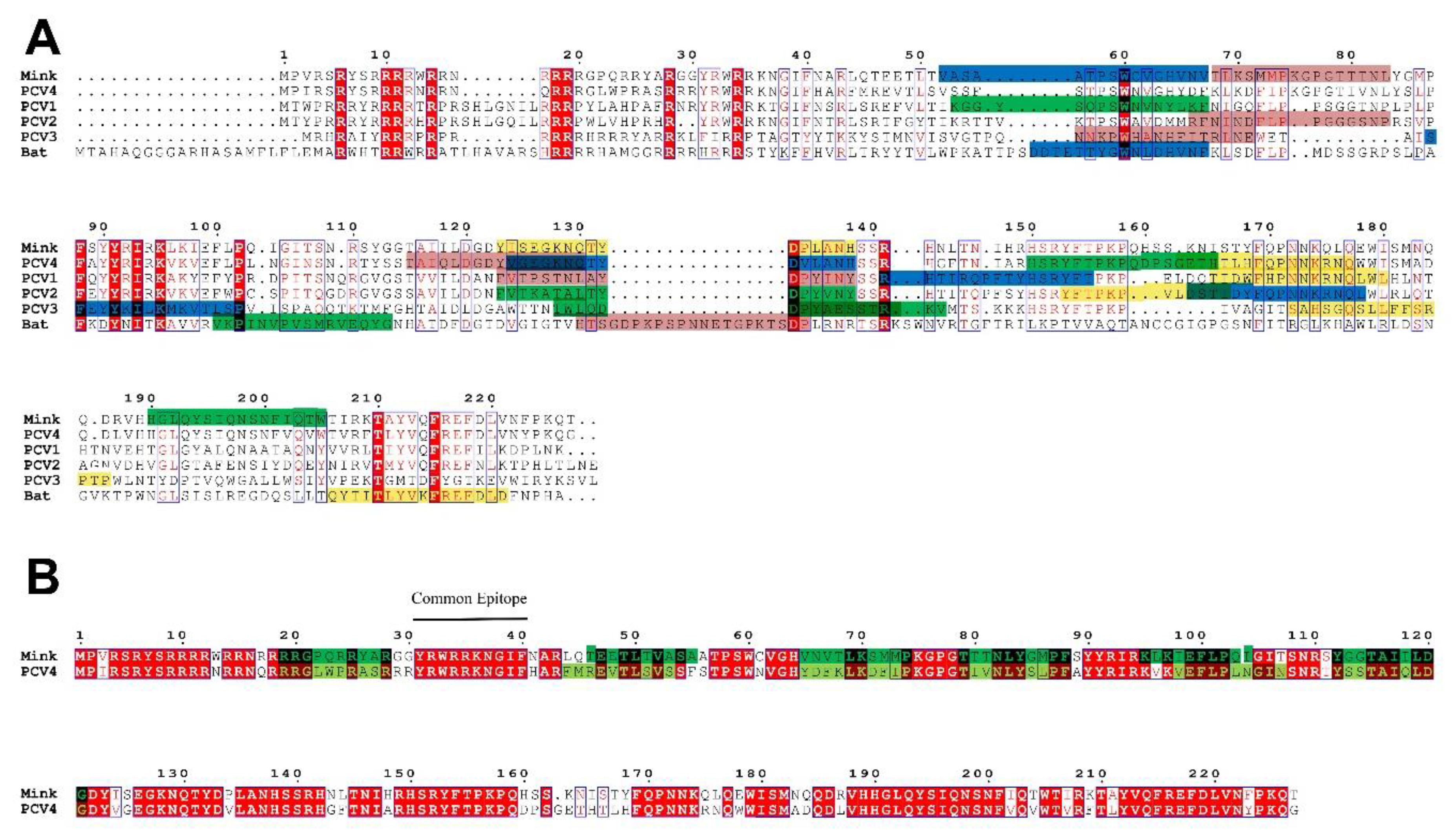
2.2. PCV4 Has Higher Homology with MiCV Than Other PCVs
2.3. The 3D Structure of Cap Protein of PCV4 Was Most Like That of MiCV
2.4. The Epitopes of PCV4 Cap Were Most Like That of MiCV
3. Discussion
4. Materials and Methods
4.1. Multiple Alignments
4.2. Phylogenetic Tree
4.3. Three-Dimensional (3D) Structures Modeling
4.4. Epitope Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ren, L.; Chen, X.; Ouyang, H. Interactions of porcine circovirus 2 with its hosts. Virus Genes 2016, 52, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, T.; Zhang, X.; Liu, X.; Ren, L. Co-infection of swine with Porcine Circovirus type 2 and other swine viruses. Viruses 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ouyang, T.; Ouyang, H.; Liu, X.; Niu, G.; Huo, W.; Yin, W.; Pang, D.; Ren, L. Human cells are permissive for the productive infection of porcine circovirus type 2 in vitro. Sci. Rep. 2019, 9, 5638. [Google Scholar] [CrossRef]
- Ouyang, T.; Niu, G.; Liu, X.; Zhang, X.; Zhang, Y.; Ren, L. Recent progress on porcine circovirus type 3. Infect. Genet. Evol. 2019, 73, 227–233. [Google Scholar] [CrossRef]
- Zhang, H.H.; Hu, W.Q.; Li, J.Y.; Liu, T.N.; Zhou, J.Y.; Opriessnig, T.; Xiao, C.T. Novel circovirus species identified in farmed pigs designated as Porcine circovirus 4, Hunan province, China. Transbound. Emerg. Dis. 2020, 67, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Hu, W.Q.; Ning, K.M.; Li, S.Y.; Xiao, C.T. The seroprevalence of the newly identified porcine circovirus type 4 in China investigated by an enzymed-linked immunosorbent assay. Transbound. Emerg. Dis. 2021, 68, 2910–2914. [Google Scholar] [CrossRef] [PubMed]
- Ha, Z.; Yu, C.; Xie, C.; Wang, G.; Zhang, Y.; Hao, P.; Li, J.; Li, Z.; Li, Y.; Rong, F.; et al. Retrospective surveillance of porcine circovirus 4 in pigs in Inner Mongolia, China, from 2016 to 2018. Arch. Virol. 2021, 166, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.G.; Do, H.Q.; Huynh, T.M.; Park, Y.H.; Park, B.K.; Chung, H.C. Molecular-based detection, genetic characterization and phylogenetic analysis of porcine circovirus 4 from Korean domestic swine farms. Transbound. Emerg. Dis. 2021. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kim, H.R.; Park, J.H.; Kwon, N.Y.; Kim, J.M.; Kim, J.K.; Park, J.H.; Lee, K.K.; Kim, S.H.; Kim, W.I.; et al. Detection of a novel porcine circovirus 4 in Korean pig herds using a loop-mediated isothermal amplification assay. J. Virol. Methods 2022, 299, 114350. [Google Scholar] [CrossRef]
- Lian, Z.; Liu, J.; Liu, P.; Zhu, Z.; Yao, X.; Yuan, L.; Hu, D.; Jiang, Y.; Chen, C.; Chen, N.; et al. Development and application of an indirect ELISA for the detection of antibody to porcine circovirus 4 in pigs. Transbound. Emerg. Dis. 2021, 68, 2975–2979. [Google Scholar] [CrossRef]
- Xu, T.; Hou, C.Y.; Zhang, Y.H.; Li, H.X.; Chen, X.M.; Pan, J.J.; Chen, H.Y. Simultaneous detection and genetic characterization of porcine circovirus 2 and 4 in Henan province of China. Gene 2022, 808, 145991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Bai, C.; Ge, K.; Li, Y.; Gao, W.; Jiang, S.; Wang, Y. Establishment of an SYBR Green-based real-time PCR assay for porcine circovirus type 4 detection. J. Virol. Methods 2020, 285, 113963. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.B.; Zhao, Y.; Cui, J.T.; Zheng, H.H.; Xu, T.; Hou, C.Y.; Wang, Z.Y.; Li, X.S.; Zheng, L.L.; Chen, H.Y. Molecular detection and phylogenetic analysis of Porcine circovirus 4 in Henan and Shanxi Provinces of China. Transbound. Emerg. Dis. 2021, 68, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Du, Q.; Han, Z.; Bi, J.; Lan, T.; Wang, W.; Zheng, M. Detection and genetic characterization of porcine circovirus 4 (PCV4) in Guangxi, China. Gene 2021, 773, 145384. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, Y.; Li, N.; Cheng, S.; Guo, L.; Zhou, Y.; Zhang, H.; Zhang, X.; Ren, L. Mink Circovirus can infect minks, foxes and raccoon dogs. Virol. Sin. 2018, 33, 561–564. [Google Scholar] [CrossRef]
- Franzo, G.; Segales, J. Porcine circovirus 2 (PCV-2) genotype update and proposal of a new genotyping methodology. PLoS ONE 2018, 13, e0208585. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, X.; Li, X.; Ouyang, T.; Niu, G.; Ouyang, H.; Ren, L. Genotyping based on complete coding sequences of porcine circovirus type 3 is stable and reliable. Infect. Genet. Evol. 2020, 78, 104116. [Google Scholar] [CrossRef]
- Wang, D.; Mai, J.; Lei, B.; Zhang, Y.; Yang, Y.; Wang, N. Structure, antigenic properties, and highly efficient assembly of PCV4 capsid protein. Front. Vet. Sci. 2021, 8, 695466. [Google Scholar] [CrossRef]
- Meng, X.J. Porcine circovirus type 2 (PCV2): Pathogenesis and interaction with the immune system. Annu. Rev. Anim. Biosci. 2013, 1, 43–64. [Google Scholar] [CrossRef]
- Palinski, R.; Pineyro, P.; Shang, P.; Yuan, F.; Guo, R.; Fang, Y.; Byers, E.; Hause, B.M. A novel porcine circovirus distantly related to known Circoviruses is associated with porcine dermatitis and nephropathy syndrome and reproductive failure. J. Virol. 2017, 91, e01879. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.G.; Giannitti, F.; Rossow, S.; Marthaler, D.; Knutson, T.P.; Li, L.; Deng, X.; Resende, T.; Vannucci, F.; Delwart, E. Detection of a novel circovirus PCV3 in pigs with cardiac and multi-systemic inflammation. Virol. J. 2016, 13, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Wang, D.; Wang, J.; Zhu, S.; She, R.; Ren, X.; Tian, J.; Quan, R.; Hou, L.; Li, Z.; et al. Induction of porcine dermatitis and nephropathy syndrome in piglets by infection with Porcine Circovirus Type 3. J. Virol. 2019, 93, e02045-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, G.; Zhang, X.; Ji, W.; Chen, S.; Li, X.; Yang, L.; Zhang, L.; Ouyang, H.; Li, C.; Ren, L. Porcine circovirus 4 rescued from an infectious clone is replicable and pathogenic in vivo. Transbound. Emerg. Dis. 2022. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Nei, A.R. A simple method for estimating and testing minimum evolution trees. Molec. Biol. Evolut. 1992, 9, 945–967. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evol. Int. J. Organ. Evol. 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucl. Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Souza, P.F.N.; Mesquita, F.P.; Amaral, J.L.; Landim, P.G.C.; Lima, K.R.P.; Costa, M.B.; Farias, I.R.; Lima, L.B.; Montenegro, R.C. The human pandemic coronaviruses on the show: The spike glycoprotein as the main actor in the coronaviruses play. Int. J. Biol. Macromol. 2021, 179, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region (Location) | Genomic Site | Mutation | |
|---|---|---|---|
| Nucleotide Acid | Amino Acid | ||
| UTR (1–74) | 72 | C-T | synonymous |
| 74 | G-A | synonymous | |
| Rep (75–965) | 84 | G-A | A4T |
| 206 | C-T | synonymous | |
| 228 | T-C | synonymous | |
| 314 | C-T | synonymous | |
| 536 | C-G/T | synonymous | |
| 537 | C-A | Q155K | |
| 599 | T-C | synonymous | |
| 608 | A-C | synonymous | |
| 641 | A-G | synonymous | |
| 726 | C-T | synonymous | |
| 752 | T-C | synonymous | |
| 757 | G-A | R228H | |
| 866 | C-T | synonymous | |
| 896 | C-T | synonymous | |
| 920 | G-A | synonymous | |
| UTR (966–1046) | 988 | C-T | synonymous |
| 1040 | C-T | synonymous | |
| Cap (complement, 1047–1733) | 1074 | A-G | synonymous |
| 1100 | T-G | M212L | |
| 1251 | T-G | synonymous | |
| 1448 | T-C | I93V | |
| 1654 | T-C | N27S | |
| 1698 | C-A | synonymous | |
| 1722 | T-C | synonymous | |
| Species | GenBank No. | PCV1 (%) | PCV2 (%) | PCV3 (%) | PCV4 (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genome | Rep Gene | Cap Gene | Genome | Rep Gene | Cap Gene | Genome | Rep Gene | Cap Gene | Genome | Rep Gene | Cap Gene | ||
| Bat | KX756986 | 23.6~24.5 | 52.7~54.4 | 23.6~24.5 | 26.8~26.0 | 53.2~54.2 | 26.8~26.0 | 17.0~17.5 | 46.0~46.4 | 17.0~17.5 | 28.5~29.0 | 52.8~53.1 | 28.5~29.0 |
| MH760364 | 50.2~51.5 | 69.4~70.7 | 50.2~51.5 | 50.7~53.5 | 71.2~71.9 | 50.7~53.5 | 28.8~29.3 | 46.0~46.7 | 28.8~29.3 | 45.7~46.2 | 47.4~48.1 | 45.7~46.2 | |
| MH760365 | 47.2~48.9 | 73.9~75.9 | 47.2~48.9 | 46.1~47.2 | 73.4~74.0 | 46.1~47.2 | 22.5~24.0 | 45.0~45.7 | 22.5~24.0 | 43.5~44.4 | 50.5~51.2 | 43.5~44.4 | |
| Mink | KJ020099 | 45.2~45.7 | 50.2~51.4 | 45.2~45.7 | 44.3~45.0 | 49.3~50.0 | 44.3~45.0 | 24.9~25.4 | 49.7~50.0 | 24.9~25.4 | 69.6~70.0 | 80.1~80.7 | 69.6~70.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Chen, S.; Niu, G.; Zhang, X.; Ji, W.; Ren, Y.; Zhang, L.; Ren, L. Porcine Circovirus Type 4 Strains Circulating in China Are Relatively Stable and Have Higher Homology with Mink Circovirus than Other Porcine Circovirus Types. Int. J. Mol. Sci. 2022, 23, 3288. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063288
Li X, Chen S, Niu G, Zhang X, Ji W, Ren Y, Zhang L, Ren L. Porcine Circovirus Type 4 Strains Circulating in China Are Relatively Stable and Have Higher Homology with Mink Circovirus than Other Porcine Circovirus Types. International Journal of Molecular Sciences. 2022; 23(6):3288. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063288
Chicago/Turabian StyleLi, Xue, Si Chen, Guyu Niu, Xinwei Zhang, Weilong Ji, Ying Ren, Liying Zhang, and Linzhu Ren. 2022. "Porcine Circovirus Type 4 Strains Circulating in China Are Relatively Stable and Have Higher Homology with Mink Circovirus than Other Porcine Circovirus Types" International Journal of Molecular Sciences 23, no. 6: 3288. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063288