
Nimodipine Exerts Time-Dependent Neuroprotective Effect after Excitotoxical Damage in Organotypic Slice Cultures

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
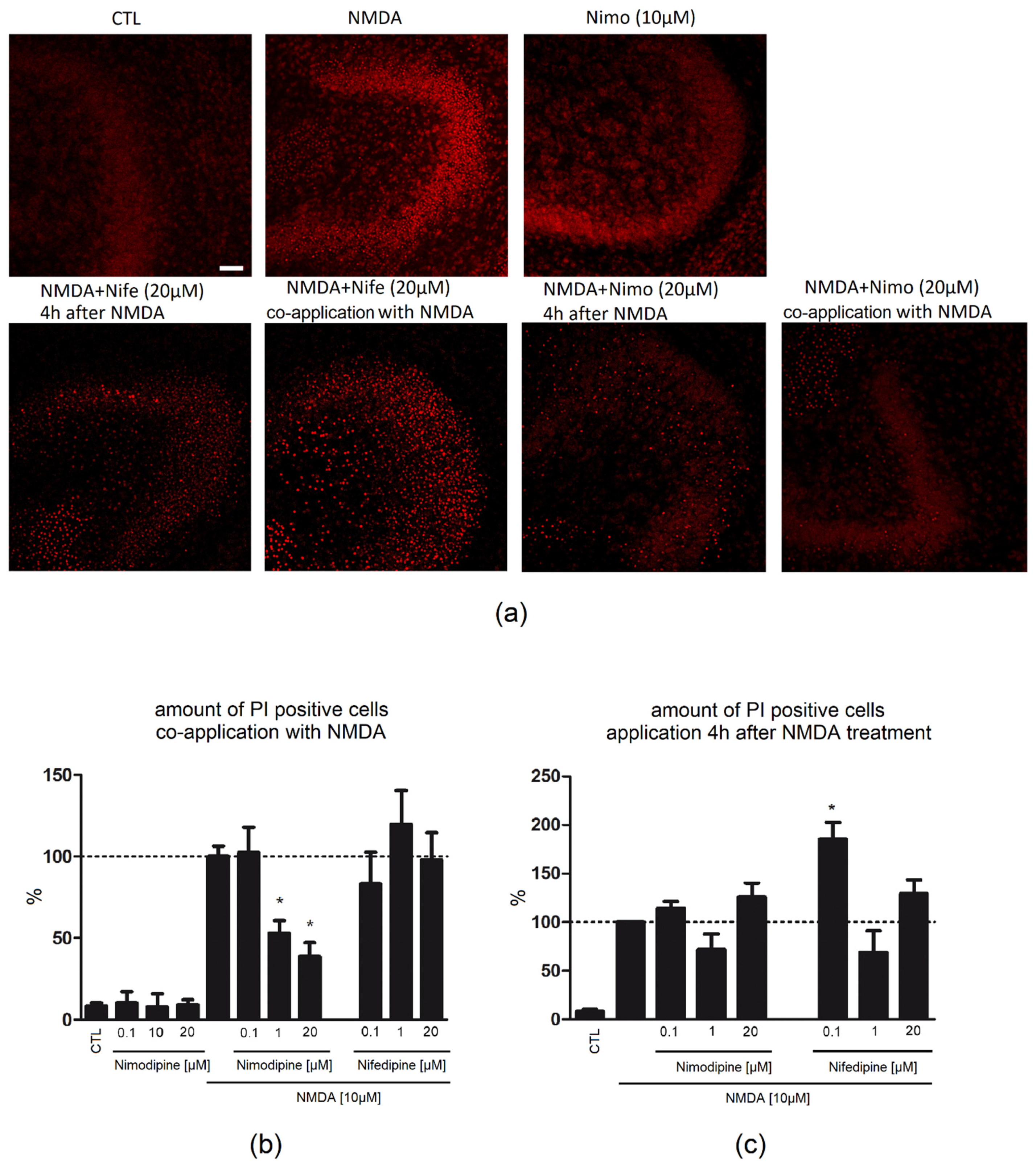
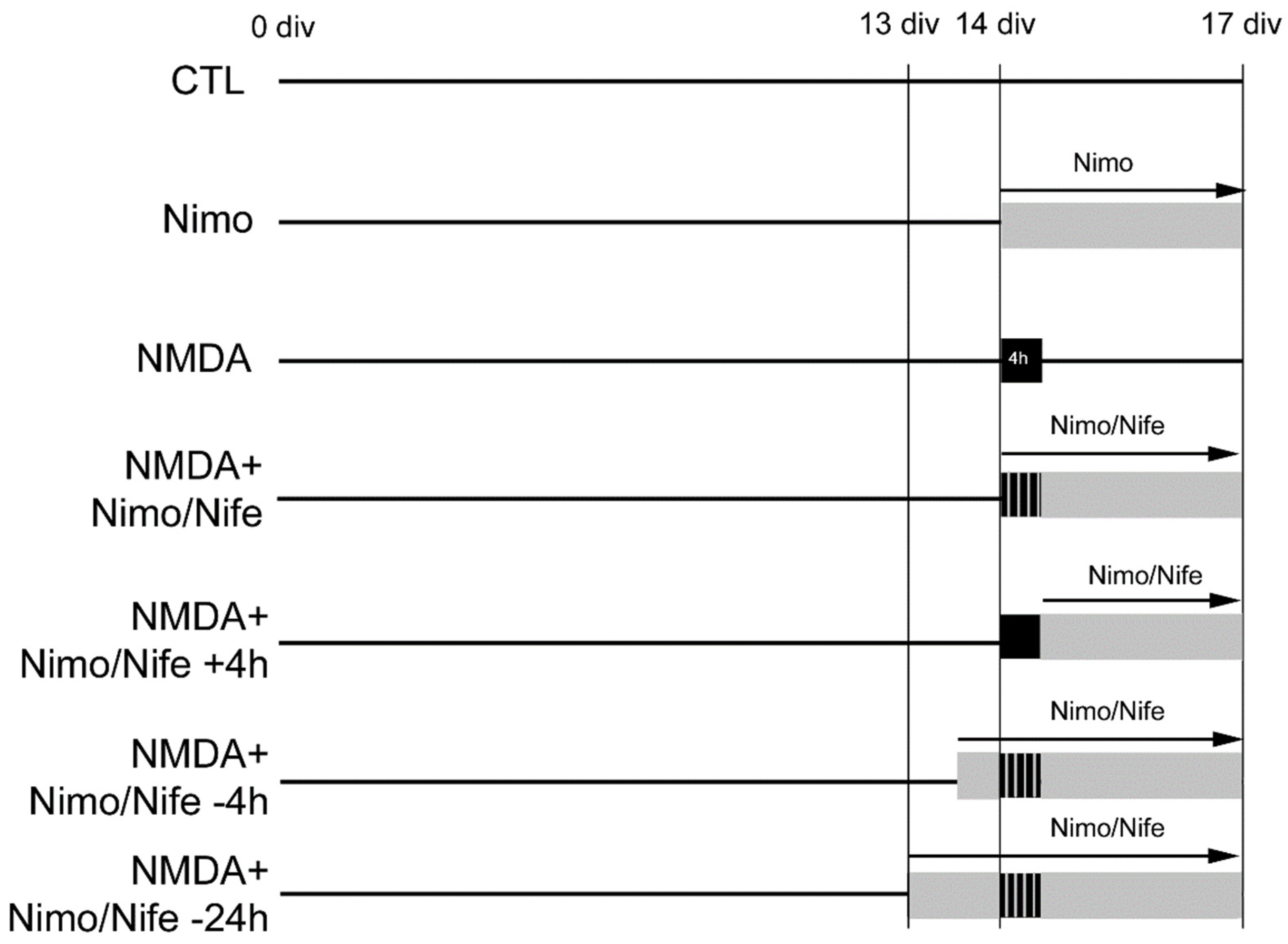
2.1. Nimodipine Is Protective When Administered Simultaneously with NMDA
2.2. Nimodipine Showed No Neuroprotective Effects When Applied after Neuronal Damage
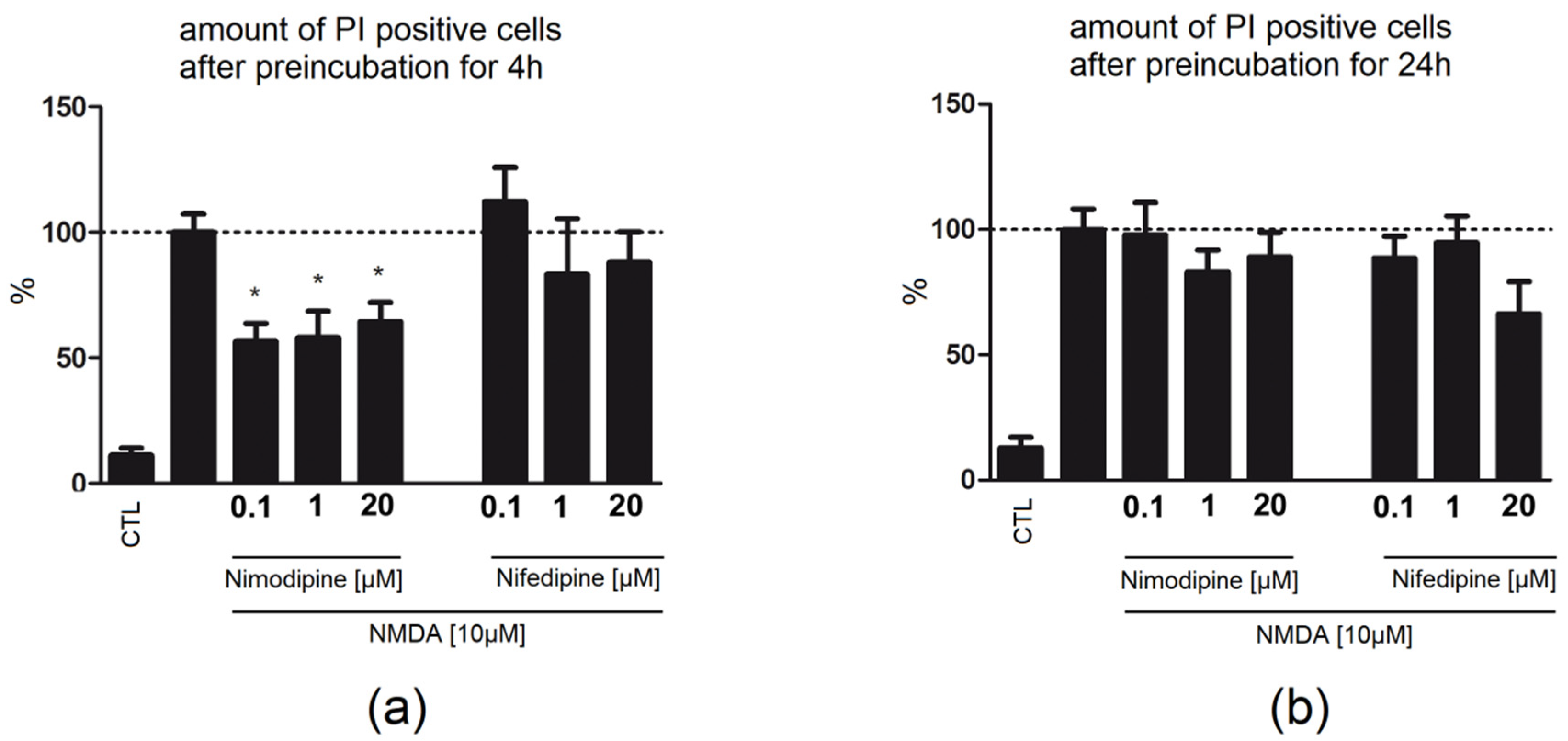
2.3. Four Hour Preincubation with Nimodipine Is Protective, Whereas 24 h Preapplication of Nimodipine or Nifedipine Had No Effect on Neuronal Damage
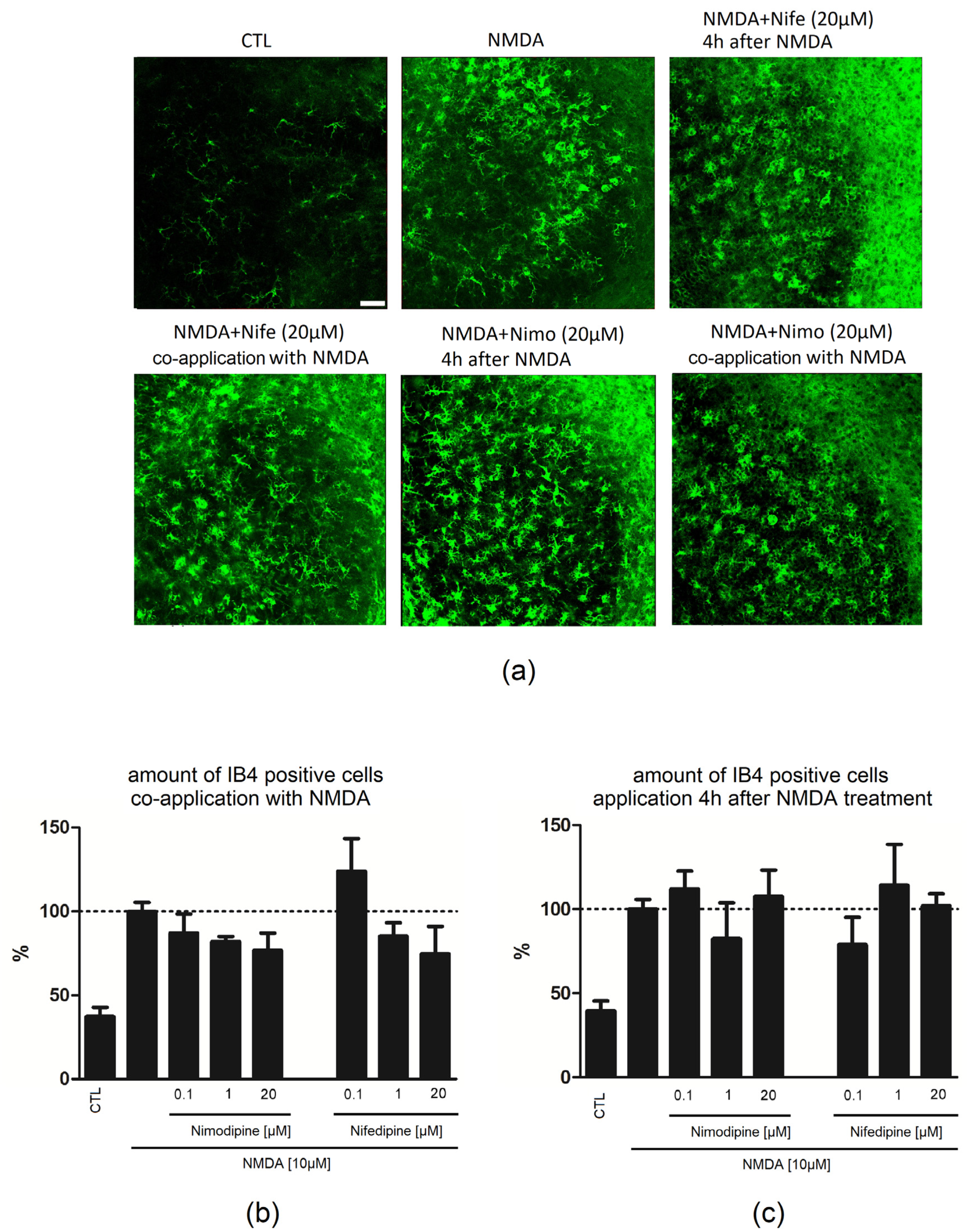
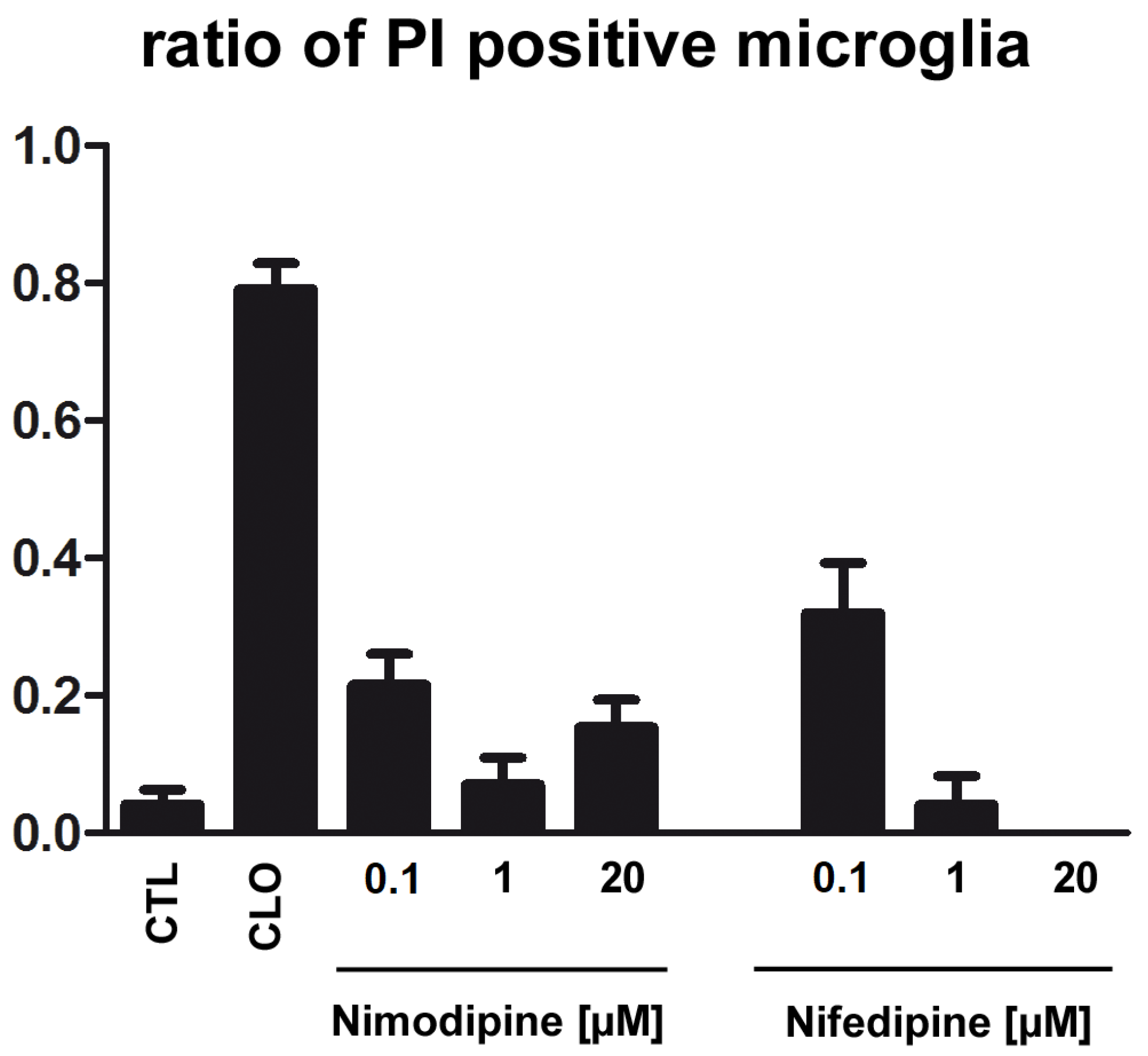
2.4. Application of Nimodipine Had No Effect on Microglia Cell Death
3. Discussion
3.1. Nimodipine but Not Nifedipine Is Protective in OHSC
3.2. The Absence of Functional Blood Vessels in OHSC and the Neuroprotective Effect of Nimodipine but Not Nifedipine Strengthens the Presence of Other Intrinsic Targets
3.3. The Application Time of Nimodipine Is Crucial
3.4. Effect of Nimodipine on Glia Cells
4. Materials and Methods
4.1. Primary Cell Cultures and Cell Lines
4.2. Organotypic Hippocampal Slice Cultures (OHSC)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meyer, F.B.; Anderson, R.E.; Sundt, T.M.; Sharbrough, F.W. Selective Central Nervous System Calcium Channel Blockers—A New Class of Anticonvulsant Agents. Mayo Clin. Proc. 1986, 61, 239–247. [Google Scholar] [CrossRef]
- Scriabine, A.; Schuurman, T.; Traber, J. Pharmacological basis for the use of nimodipine in central nervous system disorders. FASEB J. 1989, 3, 1799–1806. [Google Scholar] [CrossRef] [Green Version]
- Sandin, M.; Jasmin, S.; Levere, T. Aging and cognition: Facilitation of recent memory in aged nonhuman primates by nimodipine. Neurobiol. Aging 1990, 11, 573–575. [Google Scholar] [CrossRef]
- Horn, J.; De Haan, R.J.; Vermeulen, M.; Luiten, P.G.; Limburg, M. Nimodipine in animal model experiments of focal cerebral ischemia: A systematic review. Stroke 2001, 32, 2433–2438. [Google Scholar] [CrossRef]
- Scriabine, A.; Van den Kerckhoff, W. Pharmacology of Nimodipine—A Review. Ann. N. Y. Acad. Sci. 1988, 522, 698–706. [Google Scholar] [CrossRef]
- Carlson, A.P.; Hänggi, D.; Macdonald, R.L.; Shuttleworth, C.W. Nimodipine Reappraised: An Old Drug with a Future. Curr. Neuropharmacol. 2019, 18, 65–82. [Google Scholar] [CrossRef]
- Weiss, J.H.; Pike, C.J.; Cotman, C.W. Rapid Communication: Ca2+ Channel Blockers Attenuate β-Amyloid Peptide Toxicity to Cortical Neurons in Culture. J. Neurochem. 2008, 62, 372–375. [Google Scholar] [CrossRef]
- Fritze, J.; Walden, J. Clinical findings with nimodipine in dementia: Test of the calcium hypothesis. J. Neural Transm. Suppl. 1995, 46, 439–453. [Google Scholar]
- Moyer, J.R.; Thompson, L.; Black, J.P.; Disterhoft, J. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J. Neurophysiol. 1992, 68, 2100–2109. [Google Scholar] [CrossRef]
- Kass, I.S.; Cottrell, J.E.; Chambers, G. Magnesium and Cobalt, not Nimodipine, Protect Neurons against Anoxic Damage in the Rat Hippocampal Slice. Anesthesiology 1988, 69, 710–715. [Google Scholar] [CrossRef]
- Abele, A.E.; Scholz, K.P.; Scholz, W.K.; Miller, R.J. Excitotoxicity induced by enhanced excitatory neurotransmission in cultured hippocampal pyramidal neurons. Neuron 1990, 4, 413–419. [Google Scholar] [CrossRef]
- McLeod, J.R.; Shen, M.; Kim, D.J.; Thayer, S.A. Neurotoxicity mediated by aberrant patterns of synaptic activity between rat hippocampal neurons in culture. J. Neurophysiol. 1998, 80, 2688–2698. [Google Scholar] [CrossRef]
- Black, J.; Disterhoft, J.F.; Yeh, J.Z. Dihydropyridine effects on non-inactivating calcium currents in CA1 neurons. Soc. Neurosci. Abstr. 1990, 16, 510. [Google Scholar]
- O’Dell, T.J.; Alger, B.E. Single calcium channels in rat and guinea pig hippocampal neurons. J. Physiol. 1991, 436, 739–767. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, M.H.; Kocsis, J.D.; Waxman, S.G. Nimodipine and nifedipine enhance transmission at the Schaffer collateral CA1 pyramidal neuron synapse. Exp. Brain Res. 1991, 84, 224–228. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Liu, Y.; Bao, Y.; An, L. Nimodipine protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Neuropharmacology 2009, 56, 580–589. [Google Scholar] [CrossRef]
- Hashioka, S.; Klegeris, A.; McGeer, P.L. Inhibition of human astrocyte and microglia neurotoxicity by calcium channel blockers. Neuropharmacology 2012, 63, 685–691. [Google Scholar] [CrossRef]
- Leisz, S.; Simmermacher, S.; Prell, J.; Strauss, C.; Scheller, C. Nimodipine-dependent protection of schwann cells, astrocytes and neuronal cells from osmotic, oxidative and heat stress is associated with the activation of AKT and CREB. Int. J. Mol. Sci. 2019, 20, 4578. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Lecht, S.; Rotfeld, E.; Arien-Zakay, H.; Tabakman, R.; Matzner, H.; Yaka, R.; Lelkes, P.I.; Lazarovici, P. Neuroprotective effects of nimodipine and nifedipine in the NGF-differentiated PC12 cells exposed to oxygen-glucose deprivation or trophic withdrawal. Int. J. Dev. Neurosci. 2012, 30, 465–469. [Google Scholar] [CrossRef]
- Thompson, L.; Deyo, R.; Disterhoft, J. Nimodipine enhances spontaneous activity of hippocampal pyramidal neurons in aging rabbits at a dose that facilitates associative learning. Brain Res. 1990, 535, 119–130. [Google Scholar] [CrossRef]
- Grabiec, U.; Hohmann, T.; Hammer, N.; Dehghani, F. Organotypic Hippocampal Slice Cultures As a Model to Study Neuroprotection and Invasiveness of Tumor Cells. J. Vis. Exp. 2017, 126, e55359. [Google Scholar] [CrossRef] [PubMed]
- Murray, G.D.; Teasdale, G.M.; Schmilz, H.; Schmitz, H. Nimodipine in traumatic subarachnoid haemorrhage: A re-analysis of the HIT I and HIT II trials. Acta Neurochir. 1996, 138, 1163–1167. [Google Scholar] [CrossRef]
- Vergouwen, M.D.; Vermeulen, M.; Roos, Y.B. Effect of nimodipine on outcome in patients with traumatic subarachnoid haemorrhage: A systematic review. Lancet Neurol. 2006, 5, 1029–1032. [Google Scholar] [CrossRef]
- Macdonald, R.L. Origins of the concept of vasospasm. Stroke 2016, 47, e11–e15. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Simon, R.P.; Hallenbeck, J.M. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003, 26, 248–254. [Google Scholar] [CrossRef]
- Ricker, J.H.; Arenth, P.M. Traumatic brain injury. Funct. MRI Appl. Clin. Neurol. Psychiatry 2006, 26, 197–206. [Google Scholar]
- Mattson, M.P. Excitotoxicity. Stress Physiol. Biochem. Pathol. 2019, 3, 125–134. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, Z.-H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Hezel, M.; Koch, M.; Ghadban, C.; Korf, H.-W.; Dehghani, F. Analyses of neuronal damage in excitotoxically lesioned organotypic hippocampal slice cultures. Ann. Anat.-Anat. Anz. 2010, 192, 199–204. [Google Scholar] [CrossRef]
- Porter, N.M.; Thibault, O.; Thibault, V.; Chen, K.-C.; Landfield, P.W. Calcium Channel Density and Hippocampal Cell Death with Age in Long-Term Culture. J. Neurosci. 1997, 17, 5629–5639. [Google Scholar] [CrossRef] [Green Version]
- Brewer, L.D.; Thibault, O.; Staton, J.; Thibault, V.; Rogers, J.T.; Garcia-Ramos, G.; Kraner, S.; Landfield, P.W.; Porter, N.M. Increased vulnerability of hippocampal neurons with age in culture: Temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res. 2007, 1151, 20–31. [Google Scholar] [CrossRef]
- Regan, L.J.; Sah, D.W.; Bean, B.P. Ca2+ channels in rat central and peripheral neurons: High-threshold current resistant to dihydropyridine blockers and ω-conotoxin. Neuron 1991, 6, 269–280. [Google Scholar] [CrossRef]
- Nuglisch, J.; Karkoutly, C.; Mennel, H.D.; Roßberg, C.; Krieglstein, J. Protective Effect of Nimodipine against Ischemic Neuronal Damage in Rat Hippocampus without Changing Postischemic Cerebral Blood Flow. J. Cereb. Blood Flow Metab. 1990, 10, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Daschil, N.; Humpel, C. Nifedipine and nimodipine protect dopaminergic substantia nigra neurons against axotomy-induced cell death in rat vibrosections via modulating inflammatory responses. Brain Res. 2014, 1581, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Herzfeld, E.; Speh, L.; Strauss, C.; Scheller, C. Nimodipine but Not Nifedipine Promotes Expression of Fatty Acid 2-Hydroxylase in a Surgical Stress Model Based on Neuro2a Cells. Int. J. Mol. Sci. 2017, 18, 964. [Google Scholar] [CrossRef] [Green Version]
- Herzfeld, E.; Strauss, C.; Simmermacher, S.; Bork, K.; Horstkorte, R.; Dehghani, F.; Scheller, C. Investigation of the Neuroprotective Impact of Nimodipine on Neuro2a Cells by Means of a Surgery-Like Stress Model. Int. J. Mol. Sci. 2014, 15, 18453–18465. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Zhou, Y.; Xie, J. Nifedipine Prevents Iron Accumulation and Reverses Iron-Overload-Induced Dopamine Neuron Degeneration in the Substantia Nigra of Rats. Neurotox. Res. 2012, 22, 274–279. [Google Scholar] [CrossRef]
- Syeda, K.; Mohammed, A.M.; Arora, D.K.; Kowluru, A. Glucotoxic conditions induce endoplasmic reticulum stress to cause caspase 3 mediated lamin B degradation in pancreatic β-cells: Protection by nifedipine. Biochem. Pharmacol. 2013, 86, 1338–1346. [Google Scholar] [CrossRef] [Green Version]
- Arora, D.K.; Mohammed, A.M.; Kowluru, A. Nifedipine prevents etoposide-induced caspase-3 activation, prenyl transferase degradation and loss in cell viability in pancreatic β-cells. Apoptosis 2012, 18, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Üstün, M.; Gürbilek, M.; Ak, A.; Vatansev, H.; Duman, A. Effects of magnesium sulfate on tissue lactate and malondialdehyde levels in experimental head trauma. Intensiv. Care Med. 2001, 27, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Ina, Y.; Nagashima, K.; Ohmori, K.; Ohno, T. Antioxidant Effects of Calcium Antagonists in Rat Brain Hohogenates. Biol. Pharm. Bull. 2000, 23, 766–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramoska, E.; Spiller, H.A.; Myers, A. Calcium channel blocker toxicity. Ann. Emerg. Med. 1990, 19, 649–653. [Google Scholar] [CrossRef]
- Houston, M.C.; Olafsson, L.; Burger, M.C. Effects of Nifedipine GITS and Atenolol Monotherapy on Serum Lipis, Blood Pressure, Heart Rate, and Weight in Mild to Moderate Hypertension. Angiol. J. Vasc. Dis. 1991, 42, 681–690. [Google Scholar]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Münch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Ortner, N.J.; Striessnig, J. L-type calcium channels as drug targets in CNS disorders. Channels 2016, 10, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Latour, I.; Hamid, J.; Beedle, A.M.; Zamponi, G.W.; Macvicar, B.A. Expression of voltage-gated Ca2+ channel subtypes in cultured astrocytes. Glia 2003, 41, 347–353. [Google Scholar] [CrossRef]
- Higashi, H.; Sugita, S.; Matsunari, S.; Nishi, S. Calcium-dependent potentials with different sensitivities to calcium agonists and antagonists in guinea-pig hippocampal neurons. Neuroscience 1990, 34, 35–47. [Google Scholar] [CrossRef]
- Gähwiler, B.; Brown, D. Muscarine affects calcium-currents in rat hippocampal pyramidal cells in vitro. Neurosci. Lett. 1987, 76, 301–306. [Google Scholar] [CrossRef]
- Mattsson, P.; Aldskogius, H.; Svensson, M. Nimodipine-induced improved survival rate of facial motor neurons following intracranial transection of the facial nerve in the adult rat. J. Neurosurg. 1999, 90, 760–765. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis Is Sufficient to Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef]
- Sturniolo, R.; Altavilla, D.; Berlinghieri, M.C.; Squadrito, F.; Caputi, A.P. Splanchnic artery occlusion shock in the rat: Effects of the calcium entry blockers nimodipine and verapamil. Circ. Shock 1988, 24, 43–53. [Google Scholar]
- Zarruk, J.G.; Greenhalgh, A.D.; David, S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp. Neurol. 2018, 301, 120–132. [Google Scholar] [CrossRef]
- Espinosa-Parrilla, J.F.; Martínez-Moreno, M.; Gasull, X.; Mahy, N.; Rodríguez, M. The L-type voltage-gated calcium channel modulates microglial pro-inflammatory activity. Mol. Cell. Neurosci. 2015, 64, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Sanz, J.M.; Chiozzi, P.; Colaianna, M.; Zotti, M.; Ferrari, D.; Trabace, L.; Zuliani, G.; Di Virgilio, F. Nimodipine inhibits IL-1ß release stimulated by amyloid ß from microglia. Br. J. Pharmacol. 2012, 167, 1702–1711. [Google Scholar]
- Murakawa-Hirachi, T.; Mizoguchi, Y.; Ohgidani, M.; Haraguchi, Y.; Monji, A. Effect of memantine, an anti-Alzheimer’s drug, on rodent microglial cells in vitro. Sci. Rep. 2021, 11, 6151. [Google Scholar] [CrossRef]
- Kaindl, A.M.; Degos, V.; Peineau, S.; Gouadon, E.; Chhor, V.; Loron, G.; Le Charpentier, T.; Josserand, J.; Ali, C.; Vivien, D.; et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann. Neurol. 2012, 72, 536–549. [Google Scholar] [CrossRef]
- Wendt, S.; Wogram, E.; Korvers, L.; Kettenmann, H. Experimental Cortical Spreading Depression Induces NMDA Receptor Dependent Potassium Currents in Microglia. J. Neurosci. 2016, 36, 6165–6174. [Google Scholar] [CrossRef] [Green Version]
- Takeda, A.; Shinozaki, Y.; Kashiwagi, K.; Ohno, N.; Eto, K.; Wake, H.; Nabekura, J.; Koizumi, S. Microglia mediate non-cell-autonomous cell death of retinal ganglion cells. Glia 2018, 66, 2366–2384. [Google Scholar] [CrossRef]
- Grabiec, U.; Koch, M.; Kallendrusch, S.; Kraft, R.; Hill, K.; Merkwitz, C.; Ghadban, C.; Lutz, B.; Straiker, A.; Dehghani, F. The endocannabinoid N-arachidonoyldopamine (NADA) exerts neuroprotective effects after excitotoxic neuronal damage via cannabinoid receptor 1 (CB1). Neuropharmacology 2012, 62, 1797–1807. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Koch, M.; Pieroh, P.; Ghadban, C.; Hobusch, C.; Bechmann, I.; Dehghani, F. Time dependent neuroprotection of mycophenolate mofetil: Effects on temporal dynamics in glial proliferation, apoptosis, and scar formation. J. Neuroinflamm. 2012, 9, 89. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Hopp, S.C.; D’Angelo, H.M.; E Royer, S.; Kaercher, R.M.; Crockett, A.M.; Adzovic, L.; Wenk, G.L. Calcium dysregulation via L-type voltage-dependent calcium channels and ryanodine receptors underlies memory deficits and synaptic dysfunction during chronic neuroinflammation. J. Neuroinflamm. 2015, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Guntinas-Lichius, O.; Martinez-Portillo, F.; Lebek, J.; Angelov, D.N.; Stennert, E.; Neiss, W.F. Nimodipine maintains in vivo the increase in GFAP and enhances the astroglial ensheathment of surviving motoneurons in the rat following permanent target deprivation. J. Neurocytol. 1997, 26, 241–248. [Google Scholar] [CrossRef]
- Guo, F.; Zheng, X.; He, Z.; Zhang, R.; Zhang, S.; Wang, M.; Chen, H.; Wang, W. Nimodipine Promotes Functional Recovery after Spinal Cord Injury in Rats. Front. Pharmacol. 2021, 12, 2521. [Google Scholar] [CrossRef]
- Li, J.W.; Ren, S.H.; Ren, J.R.; Zhen, Z.G.; Li, L.R.; Hao, X.D.; Ji, H.M. Nimodipine improves cognitive impairment after subarachnoid hemorrhage in rats through incRNA NEAT1/miR-27a/MAPT axis. Drug Des. Dev. Ther. 2020, 14, 2295–2306. [Google Scholar] [CrossRef]
- Hohmann, T.; Grabiec, U.; Ghadban, C.; Feese, K.; Dehghani, F. The influence of biomechanical properties and cannabinoids on tumor invasion. Cell Adhes. Migr. 2017, 11, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Grabiec, U.; Hohmann, T.; Ghadban, C.; Rothgänger, C.; Wong, D.; Antonietti, A.; Groth, T.; Mackie, K.; Dehghani, F. Protective Effect of N-Arachidonoyl Glycine-GPR18 Signaling after Excitotoxical Lesion in Murine Organotypic Hippocampal Slice Cultures. Int. J. Mol. Sci. 2019, 20, 1266. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, U.; Pelzer, M.; Kleine, J.; Hohmann, T.; Ghadban, C.; Dehghani, F. Opposite Effects of Neuroprotective Cannabinoids, Palmitoylethanolamide, and 2-Arachidonoylglycerol on Function and Morphology of Microglia. Front. Neurosci. 2019, 13, 1180. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hohmann, U.; Ghadban, C.; Hohmann, T.; Kleine, J.; Schmidt, M.; Scheller, C.; Strauss, C.; Dehghani, F. Nimodipine Exerts Time-Dependent Neuroprotective Effect after Excitotoxical Damage in Organotypic Slice Cultures. Int. J. Mol. Sci. 2022, 23, 3331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063331
Hohmann U, Ghadban C, Hohmann T, Kleine J, Schmidt M, Scheller C, Strauss C, Dehghani F. Nimodipine Exerts Time-Dependent Neuroprotective Effect after Excitotoxical Damage in Organotypic Slice Cultures. International Journal of Molecular Sciences. 2022; 23(6):3331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063331
Chicago/Turabian StyleHohmann, Urszula, Chalid Ghadban, Tim Hohmann, Joshua Kleine, Miriam Schmidt, Christian Scheller, Christian Strauss, and Faramarz Dehghani. 2022. "Nimodipine Exerts Time-Dependent Neuroprotective Effect after Excitotoxical Damage in Organotypic Slice Cultures" International Journal of Molecular Sciences 23, no. 6: 3331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063331