
A Liquid Biopsy-Based Approach for Monitoring Treatment Response in Post-Operative Colorectal Cancer Patients


 ,
,
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
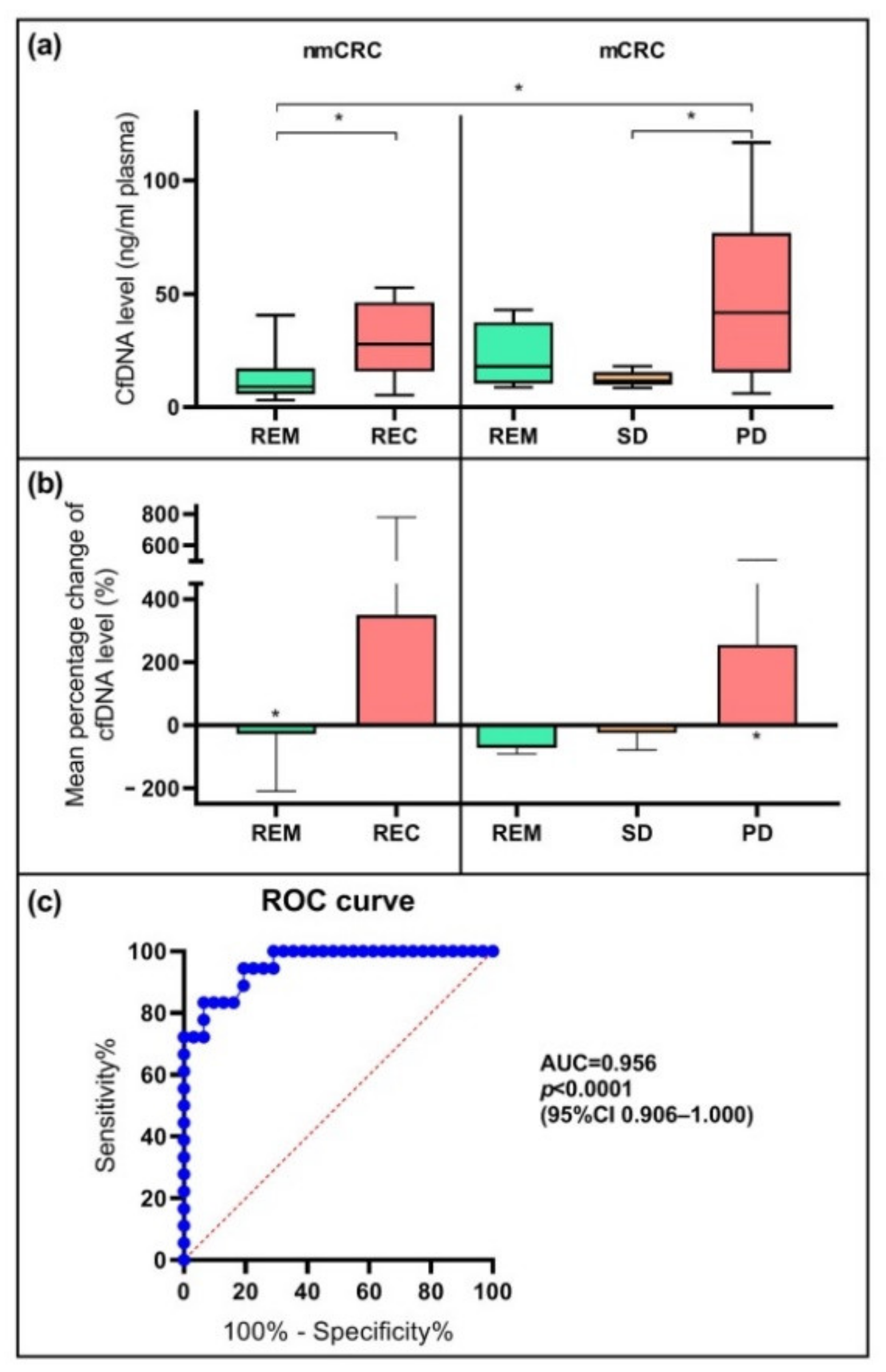
2.2. Alterations in Cell-Free DNA Level
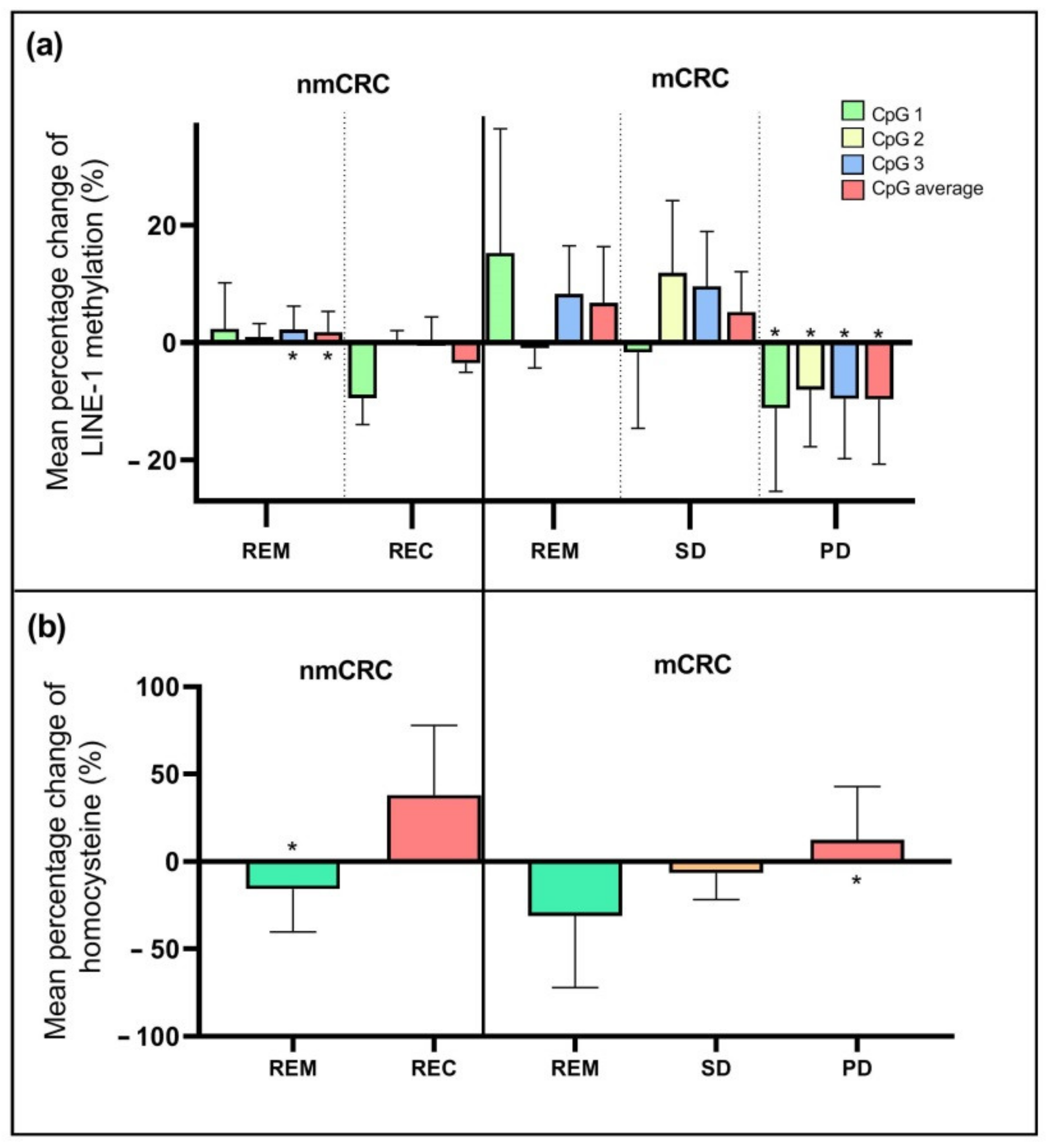
2.3. Changes in Genome-Wide DNA Methylation and Homocysteine Level
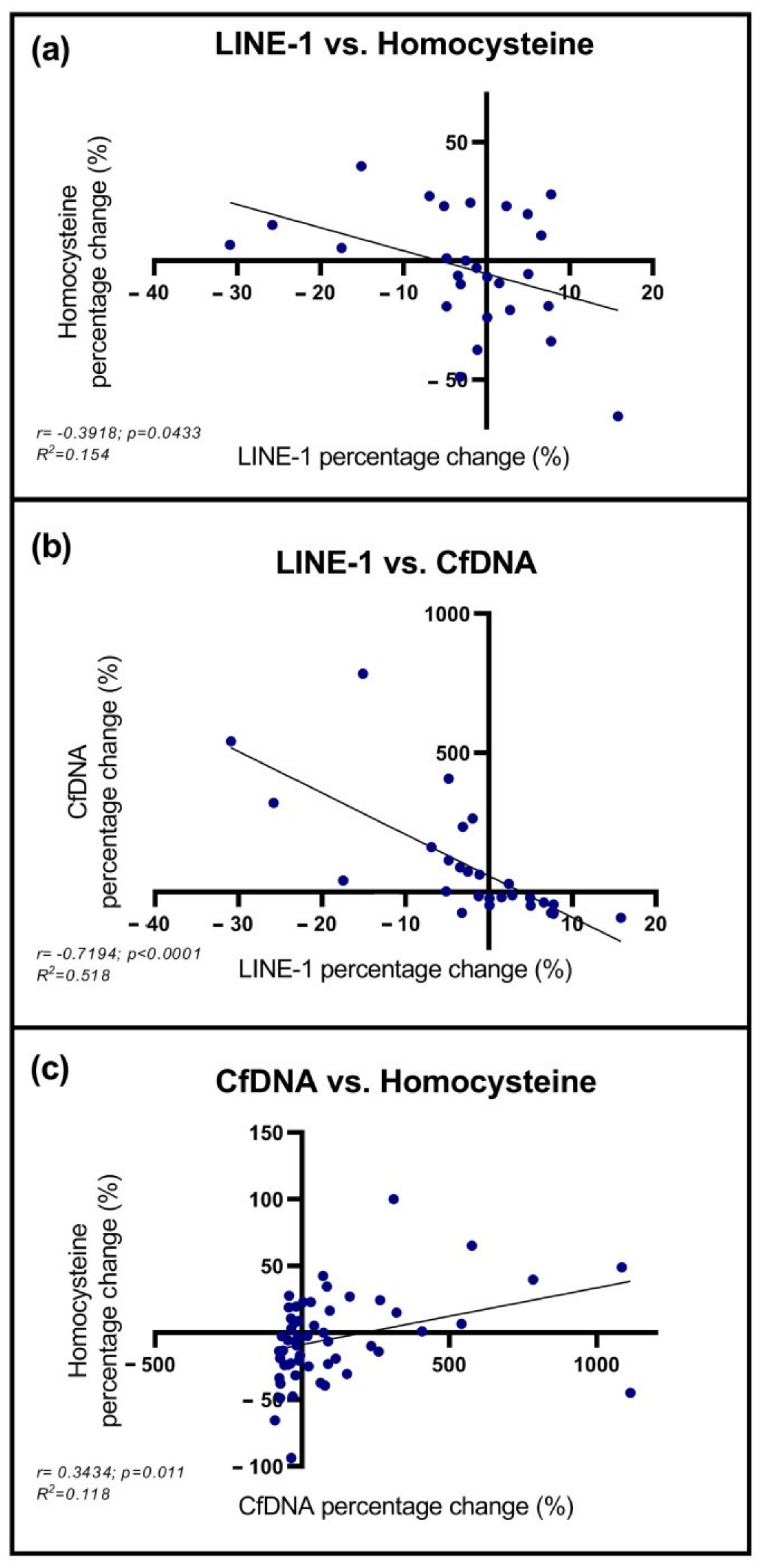
2.4. Correlation between cfDNA, LINE-1, and Homocysteine Levels
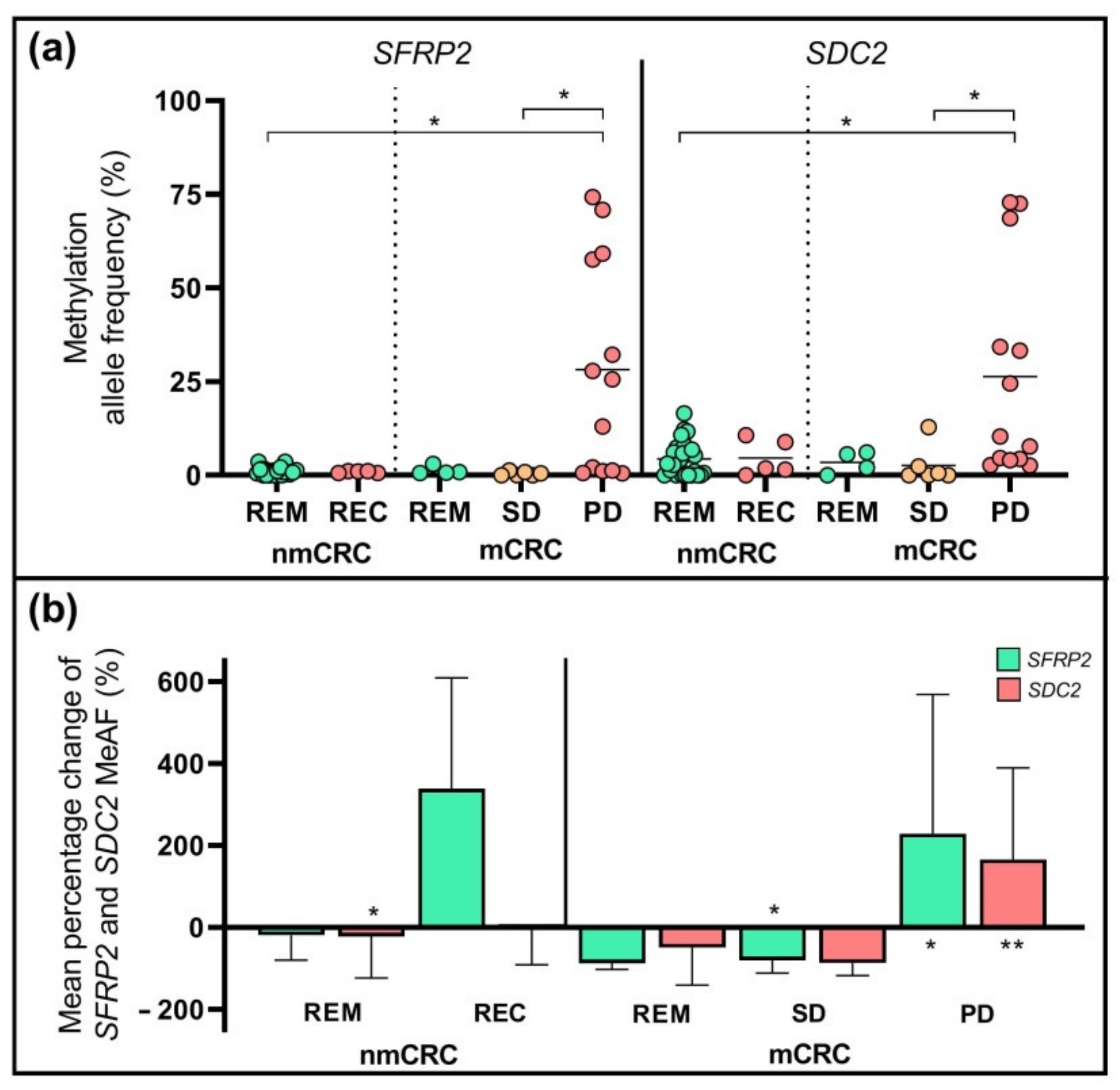
2.5. DNA Methylation Pattern of SFRP2 and SDC2 Genes
2.6. Multivariable Analysis of Selected Parameters
2.7. Analysis of CEA and CA 19-9 Tumor Markers
2.8. Association between the Analyzed Parameters and the Clinicopathological and Demographic Factors of CRC Patients
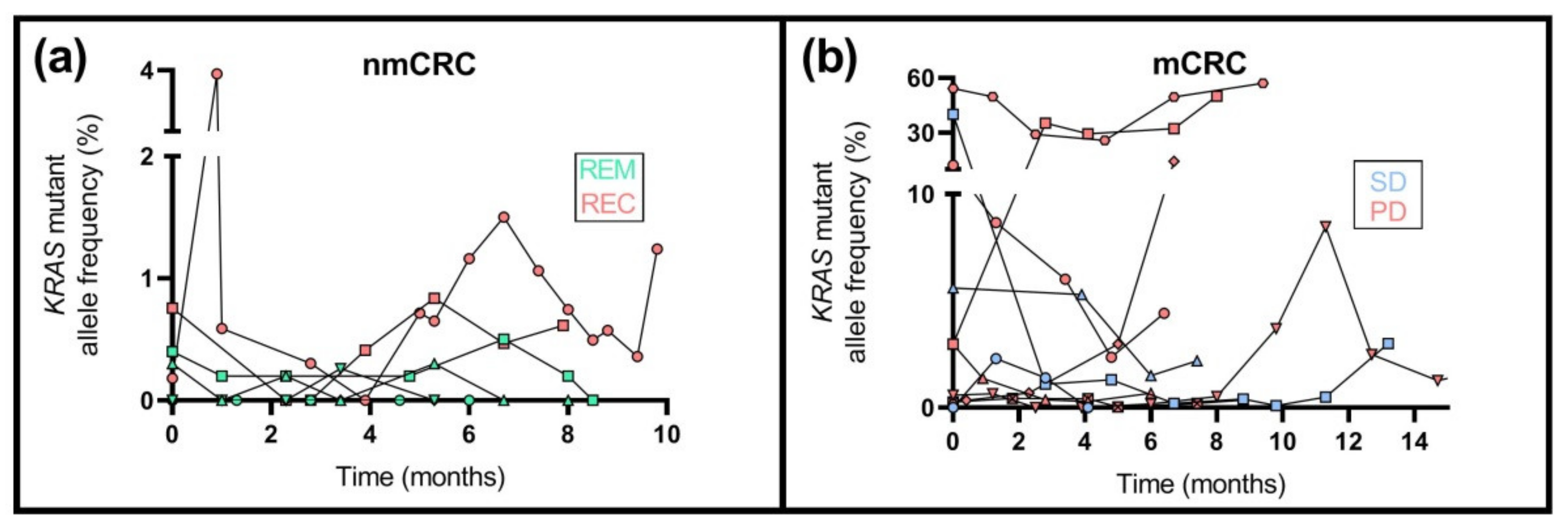
2.9. KRAS Mutation Analysis
3. Discussion
4. Materials and Methods
4.1. Patient Inclusion and Sample Collection
4.2. Cell-Free DNA Isolation and Bisulfite Conversion
4.3. Global DNA Methylation Level Analysis
4.4. SFRP2 and SDC2 Methylation Analysis
4.5. KRAS Mutation Analysis
4.6. Tumor Marker and Homocysteine Level Detection
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- McQuade, R.M.; Stojanovska, V.; Bornstein, J.C.; Nurgali, K. Colorectal Cancer Chemotherapy: The Evolution of Treatment and New Approaches. Curr. Med. Chem. 2017, 24, 1537–1557. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Prim. 2015, 1, 15065. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Verheul, H.M.; Flamen, P.; Rougier, P.; Beets-Tan, R.; Glynne-Jones, R.; Seufferlein, T. Imaging in Colorectal Cancer: Progress and Challenges for the Clinicians. Cancers 2016, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Lee, M.Y.; Park, J.H.; Park, D.I.; Sohn, C.I.; Choi, K.; Jung, Y.S. Serum CEA and CA 19-9 Levels are Associated with the Presence and Severity of Colorectal Neoplasia. Yonsei Med. J. 2017, 58, 918–924. [Google Scholar] [CrossRef]
- Hanke, B.; Riedel, C.; Lampert, S.; Happich, K.; Martus, P.; Parsch, H.; Himmler, B.; Hohenberger, W.; Hahn, E.G.; Wein, A. CEA and CA 19-9 measurement as a monitoring parameter in metastatic colorectal cancer (CRC) under palliative first-line chemotherapy with weekly 24-h infusion of high-dose 5-fluorouracil (5-FU) and folinic acid (FA). Ann. Oncol. 2001, 12, 221–226. [Google Scholar] [CrossRef]
- Wang, W.S.; Lin, J.K.; Chiou, T.J.; Liu, J.H.; Fan, F.S.; Yen, C.C.; Lin, T.C.; Jiang, J.K.; Yang, S.H.; Wang, H.S.; et al. Preoperative carcinoembryonic antigen level as an independent prognostic factor in colorectal cancer: Taiwan experience. Jpn. J. Clin. Oncol. 2000, 30, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Li, W.; Wang, K.; Xu, C.; Hao, M.; Ding, L. Perspectives of the Application of Liquid Biopsy in Colorectal Cancer. BioMed Res. Int. 2020, 2020, 6843180. [Google Scholar] [CrossRef] [Green Version]
- Barták, B.K.; Márkus, E.; Kalmár, A.; Galamb, O.; Szigeti, K.; Nagy, Z.B.; Zsigrai, S.; Tulassay, Z.; Dank, M.; Igaz, P.; et al. Characteristics and diagnostic applications of circulating cell-free DNA in colorectal cancer. Orv. Hetil. 2019, 160, 1167–1177. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Czeiger, D.; Shaked, G.; Eini, H.; Vered, I.; Belochitski, O.; Avriel, A.; Ariad, S.; Douvdevani, A. Measurement of circulating cell-free DNA levels by a new simple fluorescent test in patients with primary colorectal cancer. Am. J. Clin. Pathol. 2011, 135, 264–270. [Google Scholar] [CrossRef]
- Barault, L.; Amatu, A.; Siravegna, G.; Ponzetti, A.; Moran, S.; Cassingena, A.; Mussolin, B.; Falcomatà, C.; Binder, A.M.; Cristiano, C.; et al. Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut 2018, 67, 1995–2005. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Molnar, B.; Galamb, O.; Kalmar, A.; Bartak, B.K.; Nagy, Z.B.; Toth, K.; Tulassay, Z.; Igaz, P.; Dank, M. Circulating cell-free nucleic acids as biomarkers in colorectal cancer screening and diagnosis—An update. Expert Rev. Mol. Diagn. 2019, 19, 477–498. [Google Scholar] [CrossRef]
- Beije, N.; Helmijr, J.C.; Weerts, M.J.A.; Beaufort, C.M.; Wiggin, M.; Marziali, A.; Verhoef, C.; Sleijfer, S.; Jansen, M.; Martens, J.W.M. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol. Oncol. 2016, 10, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.K.; Lin, P.C.; Lin, C.H.; Jiang, J.K.; Yang, S.H.; Liang, W.Y.; Chen, W.S.; Chang, S.C. Clinical relevance of alterations in quantity and quality of plasma DNA in colorectal cancer patients: Based on the mutation spectra detected in primary tumors. Ann. Surg. Oncol. 2014, 21, S680–S686. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hsieh, J.S.; Chang, M.Y.; Huang, T.J.; Chen, F.M.; Cheng, T.L.; Alexandersen, K.; Huang, Y.S.; Tzou, W.S.; Lin, S.R. Molecular detection of APC, K- ras, and p53 mutations in the serum of colorectal cancer patients as circulating biomarkers. World J. Surg. 2004, 28, 721–726. [Google Scholar] [CrossRef]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [Green Version]
- Ryan, B.M.; Lefort, F.; McManus, R.; Daly, J.; Keeling, P.W.; Weir, D.G.; Kelleher, D. A prospective study of circulating mutant KRAS2 in the serum of patients with colorectal neoplasia: Strong prognostic indicator in postoperative follow up. Gut 2003, 52, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Normanno, N.; Esposito Abate, R.; Lambiase, M.; Forgione, L.; Cardone, C.; Iannaccone, A.; Sacco, A.; Rachiglio, A.M.; Martinelli, E.; Rizzi, D.; et al. RAS testing of liquid biopsy correlates with the outcome of metastatic colorectal cancer patients treated with first-line FOLFIRI plus cetuximab in the CAPRI-GOIM trial. Ann. Oncol. 2018, 29, 112–118. [Google Scholar] [CrossRef]
- Lecomte, T.; Berger, A.; Zinzindohoué, F.; Micard, S.; Landi, B.; Blons, H.; Beaune, P.; Cugnenc, P.H.; Laurent-Puig, P. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int. J. Cancer 2002, 100, 542–548. [Google Scholar] [CrossRef]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.C.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef]
- Tse, J.W.T.; Jenkins, L.J.; Chionh, F.; Mariadason, J.M. Aberrant DNA Methylation in Colorectal Cancer: What Should We Target? Trends Cancer 2017, 3, 698–712. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Kerachian, M.A.; Kerachian, M. Long interspersed nucleotide element-1 (LINE-1) methylation in colorectal cancer. Clin. Chim. Acta 2019, 488, 209–214. [Google Scholar] [CrossRef]
- Zsigrai, S.; Kalmár, A.; Nagy, Z.B.; Barták, B.K.; Valcz, G.; Szigeti, K.A.; Galamb, O.; Dankó, T.; Sebestyén, A.; Barna, G.; et al. S-Adenosylmethionine Treatment of Colorectal Cancer Cell Lines Alters DNA Methylation, DNA Repair and Tumor Progression-Related Gene Expression. Cells 2020, 9, 1864. [Google Scholar] [CrossRef]
- Wang, W.M.; Jin, H.Z. Homocysteine: A Potential Common Route for Cardiovascular Risk and DNA Methylation in Psoriasis. Chin. Med. J. Engl. 2017, 130, 1980–1986. [Google Scholar] [CrossRef]
- Mandaviya, P.R.; Stolk, L.; Heil, S.G. Homocysteine and DNA methylation: A review of animal and human literature. Mol. Genet. Metab. 2014, 113, 243–252. [Google Scholar] [CrossRef]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patai, A.V.; Molnár, B.; Kalmár, A.; Schöller, A.; Tóth, K.; Tulassay, Z. Role of DNA methylation in colorectal carcinogenesis. Dig. Dis. 2012, 30, 310–315. [Google Scholar] [CrossRef]
- deVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grutzmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin. Chem. 2009, 55, 1337–1346. [Google Scholar] [CrossRef] [Green Version]
- Imamura, Y.; Hibi, K.; Koike, M.; Fujiwara, M.; Kodera, Y.; Ito, K.; Nakao, A. RUNX3 promoter region is specifically methylated in poorly-differentiated colorectal cancer. Anticancer Res. 2005, 25, 2627–2630. [Google Scholar]
- Hanley, M.P.; Hahn, M.A.; Li, A.X.; Wu, X.; Lin, J.; Wang, J.; Choi, A.H.; Ouyang, Z.; Fong, Y.; Pfeifer, G.P.; et al. Genome-wide DNA methylation profiling reveals cancer-associated changes within early colonic neoplasia. Oncogene 2017, 36, 5035–5044. [Google Scholar] [CrossRef] [Green Version]
- Bartak, B.K.; Kalmar, A.; Peterfia, B.; Patai, A.V.; Galamb, O.; Valcz, G.; Spisak, S.; Wichmann, B.; Nagy, Z.B.; Toth, K.; et al. Colorectal adenoma and cancer detection based on altered methylation pattern of SFRP1, SFRP2, SDC2, and PRIMA1 in plasma samples. Epigenetics 2017, 12, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.W.; Schwerdel, D.; Welz, H.; Marienfeld, R.; Schmidt, S.A.; Kleger, A.; Ettrich, T.J.; Seufferlein, T. Treatment monitoring in metastatic colorectal cancer patients by quantification and KRAS genotyping of circulating cell-free DNA. PLoS ONE 2017, 12, e0174308. [Google Scholar] [CrossRef]
- Qi, J.; Qian, C.; Shi, W.; Wu, X.; Jing, R.; Zhang, L.; Wang, Z.; Ju, S. Alu-based cell-free DNA: A potential complementary biomarker for diagnosis of colorectal cancer. Clin. Biochem. 2013, 46, 64–69. [Google Scholar] [CrossRef]
- Nam, D.; Barrack, R.L.; Potter, H.G. What are the advantages and disadvantages of imaging modalities to diagnose wear-related corrosion problems? Clin. Orthop. Relat. Res. 2014, 472, 3665–3673. [Google Scholar] [CrossRef] [Green Version]
- Inanc, M.; Er, O.; Karaca, H.; Berk, V.; Ozkan, M.; Dikilitas, M.; Elmali, F. D-dimer is a marker of response to chemotherapy in patients with metastatic colorectal cancer. J. Buon 2013, 18, 391–397. [Google Scholar]
- Boysen, A.K.; Sørensen, B.S.; Lefevre, A.C.; Abrantes, R.; Johansen, J.S.; Jensen, B.V.; Schou, J.V.; Larsen, F.O.; Nielsen, D.; Taflin, H.; et al. Methodological development and biological observations of cell free DNA with a simple direct fluorescent assay in colorectal cancer. Clin. Chim. Acta 2018, 487, 107–111. [Google Scholar] [CrossRef]
- Bedin, C.; Enzo, M.V.; Del Bianco, P.; Pucciarelli, S.; Nitti, D.; Agostini, M. Diagnostic and prognostic role of cell-free DNA testing for colorectal cancer patients. Int. J. Cancer 2017, 140, 1888–1898. [Google Scholar] [CrossRef] [Green Version]
- Spindler, K.G. Methodological, biological and clinical aspects of circulating free DNA in metastatic colorectal cancer. Acta Oncol. 2017, 56, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Fittall, M.W.; Van Loo, P. Translating insights into tumor evolution to clinical practice: Promises and challenges. Genome Med. 2019, 11, 20. [Google Scholar] [CrossRef]
- Van der Pol, Y.; Mouliere, F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell 2019, 36, 350–368. [Google Scholar] [CrossRef]
- Ørntoft, M.W.; Jensen, S.; Øgaard, N.; Henriksen, T.V.; Ferm, L.; Christensen, I.J.; Reinert, T.; Larsen, O.H.; Nielsen, H.J.; Andersen, C.L. Age-stratified reference intervals unlock the clinical potential of circulating cell-free DNA as a biomarker of poor outcome for healthy individuals and patients with colorectal cancer. Int. J. Cancer 2021, 148, 1665–1675. [Google Scholar] [CrossRef]
- Nagai, Y.; Sunami, E.; Yamamoto, Y.; Hata, K.; Okada, S.; Murono, K.; Yasuda, K.; Otani, K.; Nishikawa, T.; Tanaka, T.; et al. LINE-1 hypomethylation status of circulating cell-free DNA in plasma as a biomarker for colorectal cancer. Oncotarget 2017, 8, 11906–11916. [Google Scholar] [CrossRef] [Green Version]
- Sunami, E.; de Maat, M.; Vu, A.; Turner, R.R.; Hoon, D.S. LINE-1 hypomethylation during primary colon cancer progression. PLoS ONE 2011, 6, e18884. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst. 2008, 100, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.B.; Chung, W.B.; Maeda, O.; Shin, S.J.; Kim, H.S.; Chung, H.C.; Kim, N.K.; Issa, J.P. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer 2011, 117, 1847–1854. [Google Scholar] [CrossRef] [Green Version]
- Platt, D.E.; Hariri, E.; Salameh, P.; Merhi, M.; Sabbah, N.; Helou, M.; Mouzaya, F.; Nemer, R.; Al-Sarraj, Y.; El-Shanti, H.; et al. Type II diabetes mellitus and hyperhomocysteinemia: A complex interaction. Diabetol. Metab. Syndr. 2017, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Hasan, T.; Arora, R.; Bansal, A.K.; Bhattacharya, R.; Sharma, G.S.; Singh, L.R. Disturbed homocysteine metabolism is associated with cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Ostrakhovitch, E.A.; Tabibzadeh, S. Homocysteine and age-associated disorders. Ageing Res. Rev. 2019, 49, 144–164. [Google Scholar] [CrossRef]
- Jassó, I.; Landi, A.; Dinya, E. Risk status of patients with peripheral arterial disease (PAD). Orv. Hetil. 2007, 148, 2469–2476. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.C.; Estève, P.O.; Ruse, C.I.; Pradhan, S. S-adenosylhomocysteine Hydrolase Participates in DNA Methylation Inheritance. J. Mol. Biol. 2018, 430, 2051–2065. [Google Scholar] [CrossRef]
- Pufulete, M.; Al-Ghnaniem, R.; Leather, A.J.; Appleby, P.; Gout, S.; Terry, C.; Emery, P.W.; Sanders, T.A. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: A case control study. Gastroenterology 2003, 124, 1240–1248. [Google Scholar] [CrossRef]
- Yideng, J.; Jianzhong, Z.; Ying, H.; Juan, S.; Jinge, Z.; Shenglan, W.; Xiaoqun, H.; Shuren, W. Homocysteine-mediated expression of SAHH, DNMTs, MBD2, and DNA hypomethylation potential pathogenic mechanism in VSMCs. DNA Cell Biol. 2007, 26, 603–611. [Google Scholar] [CrossRef]
- Li, J.H.; Tong, D.X.; Wang, Y.; Gao, L.; Liu, Y.; Zhang, X.H.; Chen, W.J.; Chi, J.Y.; Liu, N.; Yang, K.; et al. Neutrophil extracellular traps exacerbate coagulation and endothelial damage in patients with essential hypertension and hyperhomocysteinemia. Thromb. Res. 2021, 197, 36–43. [Google Scholar] [CrossRef]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rosales Rodriguez, I.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, Y.F.; Lu, H.N.; Wang, D.P.; Zhang, X.S.; Huang, S.L.; Sun, B.L.; Huang, Z.G. Combined detection of plasma GATA5 and SFRP2 methylation is a valid noninvasive biomarker for colorectal cancer and adenomas. World J. Gastroenterol. 2015, 21, 2629–2637. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, G.; Wang, K.; Wang, X.; Ma, Y.; Xiong, S.; Zheng, M.; Fei, S. Blood leukocytes methylation levels analysis indicate methylated plasma test is a promising tool for colorectal cancer early detection. J. Cancer 2021, 12, 3678–3685. [Google Scholar] [CrossRef] [PubMed]
- Klein-Scory, S.; Wahner, I.; Maslova, M.; Al-Sewaidi, Y.; Pohl, M.; Mika, T.; Ladigan, S.; Schroers, R.; Baraniskin, A. Evolution of RAS Mutational Status in Liquid Biopsies During First-Line Chemotherapy for Metastatic Colorectal Cancer. Front. Oncol. 2020, 10, 1115. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodriguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Pusceddu, I.; Herrmann, M.; Kirsch, S.H.; Werner, C.; Hübner, U.; Bodis, M.; Laufs, U.; Wagenpfeil, S.; Geisel, J.; Herrmann, W. Prospective study of telomere length and LINE-1 methylation in peripheral blood cells: The role of B vitamins supplementation. Eur. J. Nutr. 2016, 55, 1863–1873. [Google Scholar] [CrossRef]
- Tusnady, G.E.; Simon, I.; Varadi, A.; Aranyi, T. BiSearch: Primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res. 2005, 33, e9. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CfDNA (ng/mL Plasma) | Mean LINE-1 Methylation (%) | Homocysteine (µmol/L) | |||
|---|---|---|---|---|---|
| nmCRC | REM | Baseline | 10.1 ± 6.3 | 78.2 ± 1.3 | 13.3 ± 3.4 |
| Study end | 6.1 ± 2.8 | 80.5 ± 2.8 | 10.8 ± 3.0 | ||
| REC | Baseline | 14.9 ± 15.9 | 81.3 ± 3.3 | 10.1 ± 0.9 | |
| Study end | 32.3 ± 22.1 * | 78.5 ± 3.5 | 14.0 ± 4.1 | ||
| mCRC | REM | Baseline | 13.0 ± 5.4 | 74.7 ± 7.0 | 12.9 ± 3.9 |
| Study end | 6.3 ± 4.2 | 79.6 ± 2.0 | 7.9 ± 2.6 | ||
| SD | Baseline | 17.5 ± 13.7 | 74.7 ± 6.5 | 11.7 ± 1.6 | |
| Study end | 10.2 ± 6.0 | 78.4 ± 2.2 | 11.0 ± 2.5 | ||
| PD | Baseline | 30.6 ± 39.8 | 75.5 ± 3.4 | 10.9 ± 3.4 | |
| Study end | 75.6 ± 69.8 ** | 68.2 ± 8.4 | 13.7 ± 4.3 | ||
| Variables | Collection | Gender | Age | Tumor Location | Distant Metastasis | ||||
|---|---|---|---|---|---|---|---|---|---|
| Male | Female | <60 | >60 | Colon | Rectum | No | Yes | ||
| n = 35 | n = 20 | n = 12 | n = 43 | n = 39 | n = 16 | n = 32 | n = 23 | ||
| CfDNA | Baseline | 21.7 ± 35.8 | 14.3 ± 15.4 | 30.0 ± 45.1 | 15.9 ± 24.1 | 21.7 ± 34.6 | 12.5 ± 12.4 | 10.6 ± 11.1 | 30.6 ± 41.4 |
| (ng/mL) | Study end | 24.2 ± 41.5 | 28.9 ± 49.8 | 37.5 ± 61.1 | 22.7 ± 38.7 | 28.2 ± 49.8 | 20.4 ± 27.2 | 11.1 ± 12.7 | 46.5 ± 60.5 |
| Mean | 22.5 ± 22.9 | 22.8 ± 23.5 | 30.5 ± 25.7 | 20.4 ± 21.8 | 24.8 ± 26.0 | 17.3 ± 11.1 | 15.8 ± 12.6 | 32.1 ± 29.2 | |
| Hcy | Baseline | 12.8 ± 3.0 | 11.6 ± 3.6 | 11.5 ± 3.2 | 12.5 ± 3.4 | 12.9 ± 3.5 | 10.8 ± 2.5 | 12.8 ± 3.3 | 11.6 ± 3.2 |
| (umol/L) | Study end | 11.9 ± 3.4 | 10.0 ± 3.3 | 10.4 ± 3.8 | 11.4 ± 3.5 | 11.6 ± 3.5 | 10.3 ± 3.6 | 11.3 ± 3.3 | 11.1 ± 3.7 |
| Mean | 12.0 ± 2.6 | 9.8 ± 2.3 | 10.5 ± 3.1 | 11.6 ± 2.6 | 11.6 ± 2.9 | 10.6 ± 2.2 | 11.9 ± 2.5 | 10.6 ± 2.8 | |
| SFRP2MeAF | Baseline | 10.7 ± 22.0 | 2.4 ± 2.7 | 8.4 ± 21.6 | 7.5 ± 17.1 | 7.7 ± 16.7 | 7.6 ± 21.4 | 1.4 ± 1.6 | 16.5 ± 25.0 |
| Study end | 8.0 ± 18.4 | 6.7 ± 17.5 | 10.0 ± 21.6 | 6.8 ± 17.0 | 8.3 ± 20.1 | 5.6 ± 10.5 | 1.0 ± 0.9 | 16.3 ± 24.6 | |
| (%) | Mean | 7.2 ± 13.7 | 3.4 ± 6.0 | 6.0 ± 8.2 | 5.8 ± 12.4 | 6.5 ± 12.9 | 4.2 ± 7.6 | 1.1 ± 0.7 | 12.4 ± 15.4 |
| SDC2MeAF | Baseline | 14.5 ± 21.0 | 9.0 ± 10.6 | 12.2 ± 23.9 | 12.5 ± 16.4 | 13.2 ± 17.3 | 10.6 ± 20.3 | 8.7 ± 9.1 | 17.7 ± 24.5 |
| Study end | 11.5 ± 20.3 | 6.7 ± 6.9 | 7.1 ± 6.4 | 10.5 ± 18.7 | 10.4 ± 18.6 | 8.3 ± 11.7 | 5.1 ± 5.4 | 16.2 ± 23.4 | |
| (%) | Mean | 11.4 ± 14.8 | 8.2 ± 7.6 | 6.9 ± 6.0 | 11.2 ± 13.9 | 11.4 ± 14.2 | 7.4 ± 7.2 | 8.1 ± 7.1 | 13.2 ± 17.1 |
| CEA | Baseline | 48.4 ± 158 | 19.8 ± 55.1 | 93.1 ± 235 | 65.1 ± 268 | 55.4 ± 152 | 109 ± 422 | 1.9 ± 1.2 | 105 ± 193 |
| (ng/mL) | Study end | 27.9 ± 55.3 | 41.9 ± 138 | 73.0 ± 178 | 22.4 ± 50.6 | 37.2 ± 107 | 23.1 ± 44.4 | 2.7 ± 1.9 | 82.9 ± 135 |
| Mean | 40.9 ± 103 | 17.2 ± 31.9 | 46.3 ± 83.6 | 28.7 ± 85.9 | 58.5 ± 188 | 38.8 ± 124 | 2.5 ± 1.5 | 81.8 ± 122 | |
| CA 19-9 | Baseline | 173 ± 484 | 88.6 ± 306 | 69.0 ± 147 | 162.4 ± 475 | 150 ± 466 | 122 ± 322 | 4.8 ± 9.1 | 357 ± 617 |
| (U/mL) | Study end | 165 ± 478 | 83.1 ± 313 | 128.7 ± 404 | 136.5 ± 433 | 166 ± 488 | 60.7 ± 188 | 7.5 ± 16.3 | 335 ± 621 |
| Mean | 127 ± 356 | 46.7 ± 109 | 56.7 ± 108 | 108.6 ± 326 | 109 ± 327 | 68.0 ± 185 | 6.7 ± 11.1 | 239 ± 425 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barták, B.K.; Fodor, T.; Kalmár, A.; Nagy, Z.B.; Zsigrai, S.; Szigeti, K.A.; Valcz, G.; Igaz, P.; Dank, M.; Takács, I.; et al. A Liquid Biopsy-Based Approach for Monitoring Treatment Response in Post-Operative Colorectal Cancer Patients. Int. J. Mol. Sci. 2022, 23, 3774. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073774
Barták BK, Fodor T, Kalmár A, Nagy ZB, Zsigrai S, Szigeti KA, Valcz G, Igaz P, Dank M, Takács I, et al. A Liquid Biopsy-Based Approach for Monitoring Treatment Response in Post-Operative Colorectal Cancer Patients. International Journal of Molecular Sciences. 2022; 23(7):3774. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073774
Chicago/Turabian StyleBarták, Barbara Kinga, Tamás Fodor, Alexandra Kalmár, Zsófia Brigitta Nagy, Sára Zsigrai, Krisztina Andrea Szigeti, Gábor Valcz, Péter Igaz, Magdolna Dank, István Takács, and et al. 2022. "A Liquid Biopsy-Based Approach for Monitoring Treatment Response in Post-Operative Colorectal Cancer Patients" International Journal of Molecular Sciences 23, no. 7: 3774. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073774