
Integration of Neighbor Topologies Based on Meta-Paths and Node Attributes for Predicting Drug-Related Diseases

Abstract
:1. Introduction
- Three drug–disease heterogeneous networks were constructed, each with different aspects of drug similarities, to facilitate the acquisition of topological information regarding drug and disease nodes from different perspectives. To construct sets of different types of neighbors of the nodes, multi-scale meta-path sets of drug or disease nodes were established;
- We present an approach based on fully connected and convolutional neural networks with attention mechanisms for learning topological information regarding the same type of neighbors for drug and disease nodes. Multiple-neighbor feature representations extracted from drug and disease nodes were adaptively combined via a neighbor-scale-level attention mechanism;
- We developed a neighbor-topology-level attention mechanism to distinguish the contributions and then obtain the neighbor topological representations of the nodes; this is because different types of neighbor topological features contribute differently to drug–disease association prediction;
- The attribute information of the node pairs was extracted from the three heterogeneous networks using the proposed embedding mechanism and encoded using a convolutional autoencoder (CAE). The premise of this embedding mechanism is that drug–disease pairs are more likely to be associated with each other if they exhibit similarities or associations with more typical drugs or diseases.
2. Experimental Results and Discussion
2.1. Evaluation Metrics
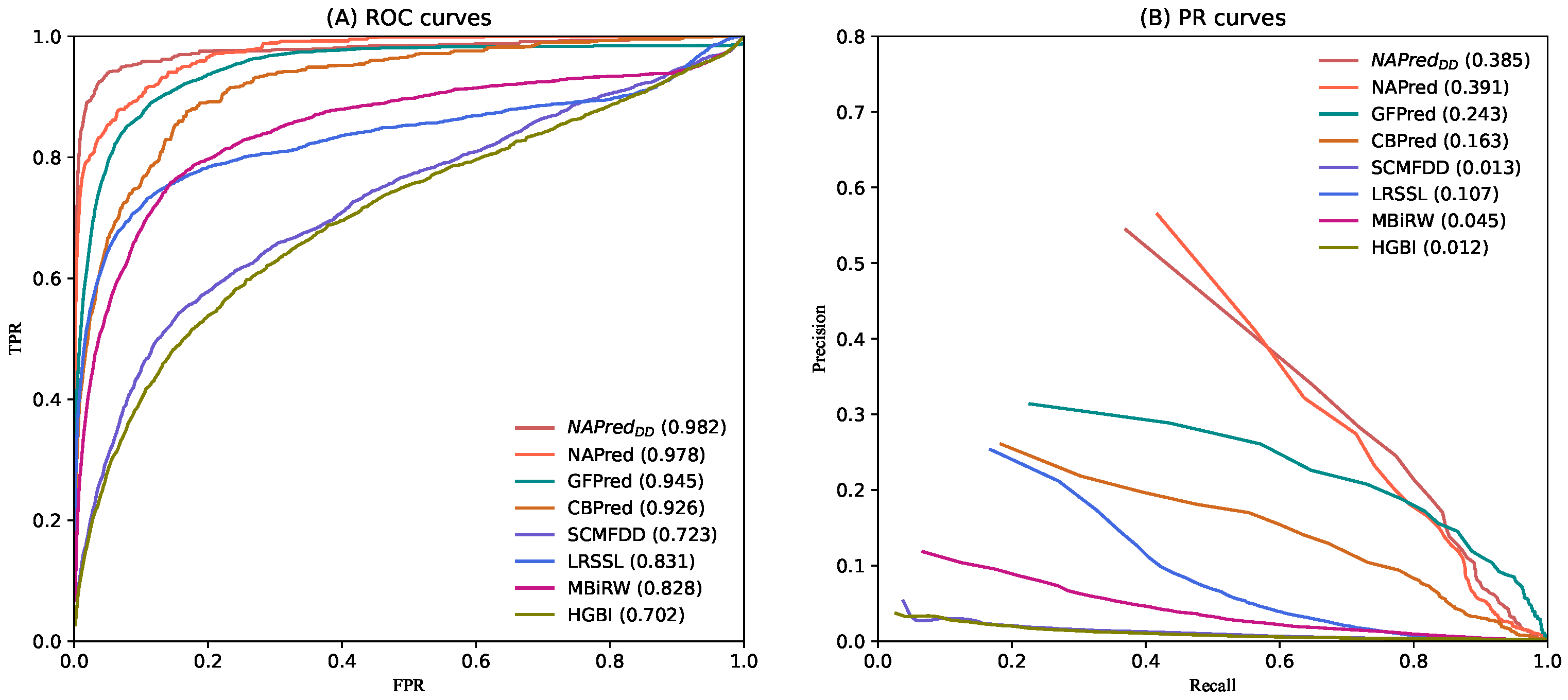
2.2. Comparison with Other Methods
2.3. Case Studies of Five Drugs
2.4. Prediction of Novel Drug-Related Diseases
3. Materials and Methods
3.1. Dataset
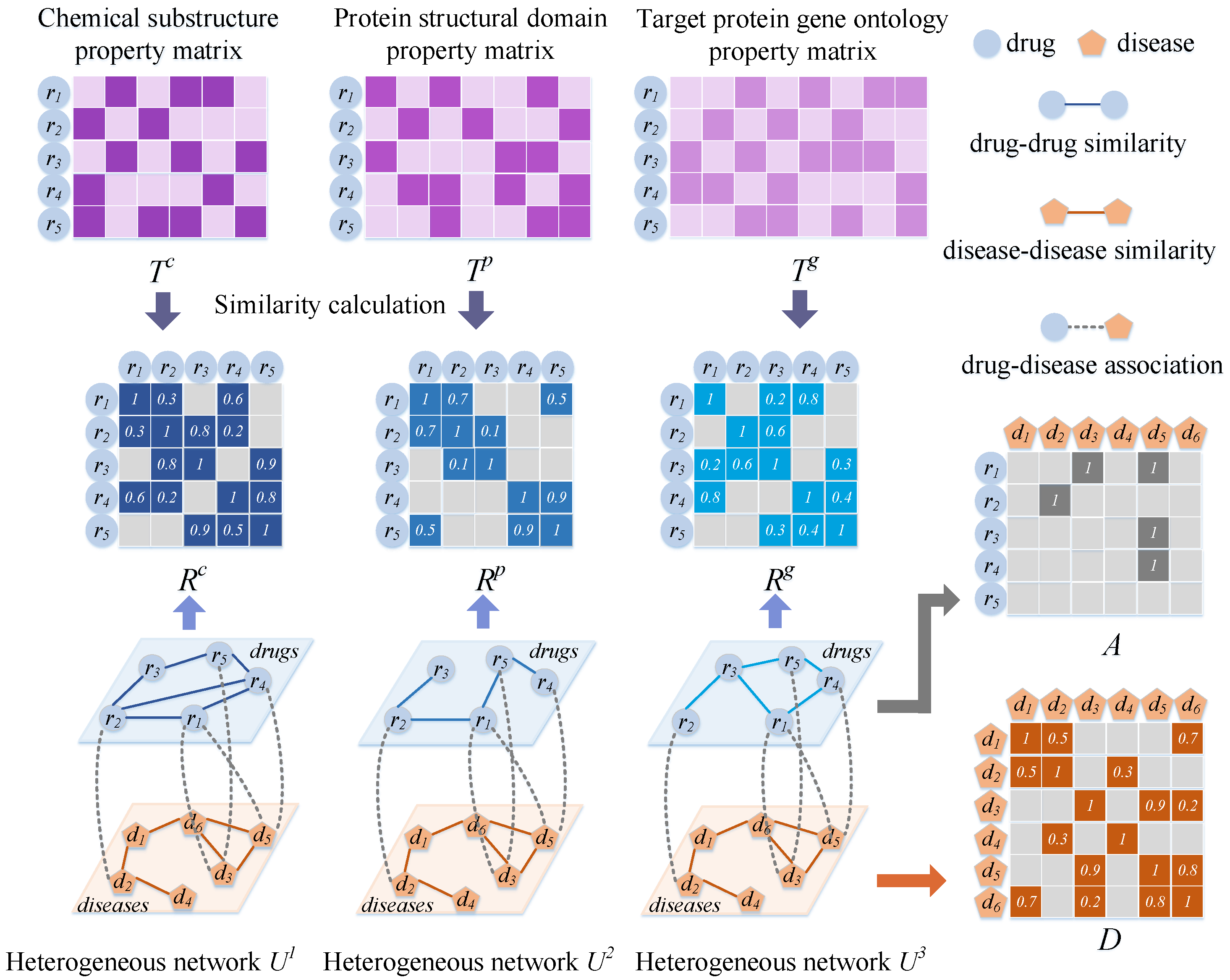
3.2. Establishing Drug–Disease Heterogeneous Networks
3.2.1. Matrix of Drug Properties
3.2.2. Establishment of the Drug Network
3.2.3. Establishment of the Disease Network
3.2.4. Drug–Disease Heterogeneous Network
3.3. Neighborhood Topology Encoding
3.3.1. Multi-Scale Meta-Path Sets
3.3.2. Neighbor Sets Based on Meta-Paths at Different Scales
3.3.3. Aggregation of Multi-Scale Neighbor Features
3.3.4. Same-Type Neighbor Topology Encoding Based on Neighbor-Scale-Level Attention
3.3.5. Neighbor Topology Encoding Based on Attention Enhancement at the Neighbor Topology Level
3.3.6. CNN-Based Pairwise Neighbor Topology Encoding
3.4. Encoding Pairwise Node Attributes
3.4.1. Attribute Embedding Matrix for Drug–Disease Pairs
3.4.2. CAE-Based Pairwise Node Attribute Encoding
3.5. Final Integration and Optimization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, H.; Cheng, F.; Li, J. iDrug: Integration of drug repositioning and drug-target prediction via cross-network embedding. PLoS Comput. Biol. 2020, 16, e1008040. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, G.; Pinoli, P.; Ceri, S.; Masseroli, M. Matrix Factorization-based Technique for Drug Repurposing Predictions. IEEE J. Biomed. Health Inform. 2020, 24, 3162–3172. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, M.; Yang, M.; Wu, F.X.; Li, Y.; Wang, J. Biomedical data and computational models for drug repositioning: A comprehensive review. Briefings Bioinform. 2021, 22, 1604–1619. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Andrew, D.; Tim, G.; Joanna, L.; Christine, M.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Turanli, B.; Altay, O.; Borén, J.; Hasan, T.; Jens, N.; Mathias, U.; Yalcin, A.K.; Adil, M. Systems biology based drug repositioning for development of cancer therapy. Semin. Cancer Biol. 2021, 68, 47–58. [Google Scholar] [CrossRef]
- Padhy, B.; Gupta, Y. Drug repositioning: Re-investigating existing drugs for new therapeutic indications. J. Postgrad. Med. 2011, 57, 153–160. [Google Scholar] [CrossRef]
- Pritchard, J.-L.E.; O’Mara, T.A.; Glubb, D.M. Enhancing the Promise of Drug Repositioning through Genetics. Front. Pharmacol. 2017, 8, 896. [Google Scholar] [CrossRef] [Green Version]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef]
- Alfedi, G.; Luffarelli, R.; Condò, I.; Pedini, G.; Mannucci, L.; Massaro, D.S. Drug repositioning screening identifies etravirine as a potential therapeutic for friedreich’s ataxia. Mov. Disord. 2019, 34, 323–334. [Google Scholar] [CrossRef]
- Karaman, B.; Sippl, W. Computational Drug Repurposing: Current Trends. Curr. Med. Chem. 2019, 26, 5389–5409. [Google Scholar] [CrossRef]
- Shameer, K.; Readhead, B.; Dudley, J.T. Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr. Top. Med. Chem. 2015, 15, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Sharan, R. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011, 7, 496. [Google Scholar] [CrossRef]
- Zhang, W.; Yue, X.; Lin, W.; Wu, W.; Liu, R.; Huang, F.; Liu, F. Predicting drug-disease associations by using similarity constrained matrix factorization. BMC Bioinform. 2018, 19, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chen, S.; Deng, N.; Wang, Y. Drug Repositioning by Kernel-Based Integration of Molecular Structure, Molecular Activity, and Phenotype Data. PLoS ONE 2013, 8, e78518. [Google Scholar]
- Liang, X.; Zhang, P.; Yan, L.; Fu, Y.; Peng, F.; Qu, L. LRSSL: Predict and interpret drug–disease associations based on data integration using sparse subspace learning. Bioinformatics 2017, 33, 1187–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WWang, W.; Yang, S.; Zhang, X.; Li, J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014, 30, 2923–2930. [Google Scholar] [CrossRef]
- Liu, H.; Song, Y.; Guan, J.; Luo, L.; Zhuang, Z. Inferring new indications for approved drugs via random walk on drug-disease heterogenous networks. BMC Bioinform. 2016, 17, 539. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef]
- Yu, L.; Su, R.; Wang, B.; Zhang, L.; Zou, Y.; Zhang, J.; Gao, L. Prediction of Novel Drugs for Hepatocellular Carcinoma Based on Multi-Source Random Walk. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 966–977. [Google Scholar] [CrossRef]
- Huang, Y.-F.; Yeh, H.-Y.; Soo, V.-W. Inferring drug-disease associations from integration of chemical, genomic and phenotype data using network propagation. BMC Med. Genom. 2013, 6, S4. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, Z.; Peng, W. miRDDCR: A miRNA-based method to comprehensively infer drug-disease causal relationships. Sci. Rep. 2017, 7, 15921. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Zhang, Y.; Zhang, T.; Li, L.; Zhao, L. Predicting MiRNA-Disease Associations by Incorporating Projections in Low-Dimensional Space and Local Topological Information. Genes 2019, 10, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, P.; Pan, S.; Zhang, T.; Liu, Y.; Sun, H. Graph Convolutional Network and Convolutional Neural Network Based Method for Predicting LncRNA-Disease Associations. Cells 2019, 8, 1012. [Google Scholar] [CrossRef] [Green Version]
- Xuan, P.; Sheng, N.; Zhang, T.; Liu, Y.; Guo, Y. CNNDLP: A Method Based on Convolutional Autoencoder and Convolutional Neural Network with Adjacent Edge Attention for Predicting LncRNA–Disease Associations. Int. J. Mol. Sci. 2019, 20, 4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, P.; Gao, L.; Sheng, N.; Zhang, T.; Nakaguchi, T. Graph Convolutional Autoencoder and Fully-Connected Autoencoder with Attention Mechanism Based Method for Predicting Drug-Disease Associations. IEEE J. Biomed. Health Inform. 2021, 25, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Ye, Y.; Zhang, T.; Zhao, L.; Sun, C. Convolutional Neural Network and Bidirectional Long Short-Term Memory-Based Method for Predicting Drug–Disease Associations. Cells 2019, 8, 705. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.-J.; Huang, Y.-A.; You, Z.-H. Predicting Drug-Disease Associations via Using Gaussian Interaction Profile and Kernel-Based Autoencoder. Biomed Res. Int. 2019, 2019, 2426958. [Google Scholar] [CrossRef]
- Hajian-Tilaki, K. Receiver Operating Characteristic (ROC) Curve Analysis for Medical Diagnostic Test Evaluation. Casp. J. Intern. Med. 2013, 4, 627–635. [Google Scholar]
- Saito, T.; Rehmsmeier, M. The Precision-Recall Plot Is More Informative than the ROC Plot When Evaluating Binary Classifiers on Imbalanced Datasets. PLoS ONE 2015, 10, e0118432. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.X.; Huang, J.; Zhang, H. AUC: A Better Measure than Accuracy in Comparing Learning Algorithms. In Conference of the Canadian Society for Computational Studies of Intelligence; Springer: Berlin/Heidelberg, Germany, 2003; pp. 329–341. [Google Scholar]
- Bolboacă, S.D.; Jäntschi, L. Predictivity Approach for Quantitative Structure-Property Models. Application for Blood-Brain Barrier Permeation of Diverse Drug-Like Compounds. Int. J. Mol. Sci. 2011, 12, 4348. [Google Scholar] [CrossRef] [Green Version]
- Bolboacă, S.D.; Jäntschi, L. Sensitivity, Specificity, and Accuracy of Predictive Models on Phenols Toxicity. J. Comput. Sci. 2014, 5, 345–350. [Google Scholar] [CrossRef]
- Pahikkala, T.; Airola, A.; Pietilä, S.; Shakyawar, S.; Szwajda, A.; Tang, J.; Aittokallio, T. Toward more realistic drug–target interaction predictions. Briefings Bioinform. 2015, 16, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Duran-Frigola, M.; Fernández-Torras, A.; Bertoni, M.; Aloy, P. Formatting biological big data for modern machine learning in drug discovery. WIREs Comput. Mol. Sci. 2019, 9, e1408. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J. The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, P.; Cao, N.; Hu, J.; Sorrentino, R. Exploring the associations between drug side-effects and therapeutic indications. J. Biomed. Inform. 2014, 51, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; Craig, M.; Conor, M.; Gift, N.; Sebastien, P.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef]
- The UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38, D142–D148. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ji, H.; Shi, C.; Wang, B.; Ye, Y.; Cui, P.; Yu, P.S. Heterogeneous Graph Attention Network. arXiv 2019, arXiv:1903.07293. [Google Scholar]
- Cen, Y.; Zou, X.; Zhang, J.; Yang, H.; Zhou, J.; Tang, J. Representation Learning for Attributed Multiplex Heterogeneous Network. arXiv 2019, arXiv:1905.01669. [Google Scholar]
- Nair, V.; Hinton, G.E. Rectified Linear Units Improve Restricted Boltzmann Machines. In Proceedings of the 27th International Conference on International Conference on Machine Learning, Haifa, Israel, 21–24 June 2010; pp. 807–814. [Google Scholar]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural Machine Translation by Jointly Learning to Align and Translate. arXiv 2014, arXiv:1409.0473. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Petrini, M. Improvements to the Backpropagation Algorithm. Ann. Univ. Petrosani Econ. 2012, 12, 185–192. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GFPred | CBPred | SCMFDD | LRSSL | MBiRW | HGBI | |
|---|---|---|---|---|---|---|
| p-value of AUC | 5.27051 × 10 | 1.83480 × 10 | 5.49787 × 10 | 5.31080 × 10 | 2.89205 × 10 | 1.74747 × 10 |
| p-value of AUCPR | 3.42304 × 10 | 4.72506 × 10 | 1.81013 × 10 | 8.63715 × 10 | 4.68094 × 10 | 4.85712 × 10 |
| Drug Name | Rank | Disease Name | Description | Rank | Disease Name | Description |
|---|---|---|---|---|---|---|
| 1 | Staphylococcal Infections | CTD, PubChem | 6 | Staphylococcal Skin | PubChem | |
| Infections | ||||||
| 2 | Pneumonia, Bacterial | ClinicalTrials | 7 | Streptococcal Infections | CTD, ClinicalTrials | |
| Ampicillin | 3 | Urinary Tract Infections | CTD, DrugBank, | 8 | Osteomyelitis | PubChem, |
| PubChem | ClinicalTrials | |||||
| 4 | Wound Infection | PubChem, ClinicalTrials | 9 | Postoperative Complications | PubChem | |
| 5 | Proteus Infections | Inferred Candidate | 10 | Bacterial Infections | CTD, DrugBank, | |
| by 2 Literature Works | ClinicalTrials | |||||
| 1 | Escherichia coli Infections | CTD, PubChem, ClinicalTrials | 6 | Salmonella Infections | DrugBank, PubChem, ClinicalTrials | |
| 2 | Urinary Tract Infections | DrugBank, PubChem, | 7 | Enterobacteriaceae Infections | PubChem, ClinicalTrials | |
| ClinicalTrials | ||||||
| Ceftriaxone | 3 | Haemophilus Infections | PubChem | 8 | Septicemia | DrugBank, PubChem, |
| ClinicalTrials | ||||||
| 4 | Gonorrhea | DrugBank, PubChem, | 9 | Endocarditis, Bacterial | DrugBank, ClinicalTrials | |
| ClinicalTrials | ||||||
| 5 | Gram-Negative Bacterial | Inferred Candidate | 10 | Pseudomonas Infections | PubChem | |
| Infections | by 1 Literature Work | |||||
| 1 | Urinary Tract Infections | CTD, PubChem | 6 | Leukemia, Lymphoid | CTD, DrugBank, | |
| ClinicalTrials | ||||||
| 2 | Leukemia, Myeloid, | CTD, DrugBank, | 7 | Bronchitis | CTD | |
| Acute | ClinicalTrials | |||||
| Doxorubicin | 3 | Escherichia coli Infections | CTD | 8 | Sarcoma | CTD, DrugBank, |
| ClinicalTrials | ||||||
| 4 | Neoplasms | ClinicalTrials, PubChem | 9 | Gonorrhea | Unconfirmed | |
| 5 | Staphylococcal Infections | CTD, PubChem | 10 | Precursor Cell Lymphoblastic | CTD | |
| Leukemia-Lymphoma | ||||||
| 1 | Gonorrhea | DrugBank, PubChem | 6 | Gram-Positive Bacterial Infections | PubChem | |
| 2 | Gram-Negative Bacterial | PubChem | 7 | Staphylococcal Infections | CTD, DrugBank, | |
| Erythromycin | Infections | PubChem | ||||
| 3 | Chancroid | DrugBank, PubChem | 8 | Pneumonia, Mycoplasma | Unconfirmed | |
| 4 | Bacterial Infections | DrugBank, PubChem | 9 | Neurosyphilis | PubChem | |
| 5 | Neisseriaceae Infections | DrugBank | 10 | Chlamydiaceae Infections | DrugBank, ClinicalTrials | |
| 1 | Candidiasis, Cutaneous | DrugBank, PubChem, | 6 | Tinea Capitis | DrugBank, PubChem | |
| ClinicalTrials | ||||||
| 2 | Tinea Versicolor | DrugBank, PubChem, | 7 | Fungemia | DrugBank, PubChem, | |
| ClinicalTrials | ClinicalTrials | |||||
| Itraconazole | 3 | Tinea Pedis | DrugBank, PubChem | 8 | Skin Diseases, Infectious | PubChem, ClinicalTrials |
| 4 | Leishmaniasis | CTD, PubChem, | 9 | AIDS-Related Opportunistic | ClinicalTrials | |
| ClinicalTrials | Infections | |||||
| 5 | Chromoblastomycosis | DrugBank, PubChem | 10 | Candidiasis | CTD, DrugBank, PubChem |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, P.; Lu, Z.; Zhang, T.; Liu, Y.; Nakaguchi, T. Integration of Neighbor Topologies Based on Meta-Paths and Node Attributes for Predicting Drug-Related Diseases. Int. J. Mol. Sci. 2022, 23, 3870. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073870
Xuan P, Lu Z, Zhang T, Liu Y, Nakaguchi T. Integration of Neighbor Topologies Based on Meta-Paths and Node Attributes for Predicting Drug-Related Diseases. International Journal of Molecular Sciences. 2022; 23(7):3870. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073870
Chicago/Turabian StyleXuan, Ping, Zixuan Lu, Tiangang Zhang, Yong Liu, and Toshiya Nakaguchi. 2022. "Integration of Neighbor Topologies Based on Meta-Paths and Node Attributes for Predicting Drug-Related Diseases" International Journal of Molecular Sciences 23, no. 7: 3870. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073870