1. Introduction
Intracellular trafficking of lipids is an essential event for the metabolism of lipids and membrane biogenesis in cells. In eukaryotic cells, the endoplasmic reticulum (ER) is the center of de novo synthesis of various lipid types, and lipid transport mediated by lipid transfer proteins (LTPs) at organelle membrane contact sites (MCSs) is the predominant mechanism to transport lipid from the ER to other organelles [
1,
2]. However, how the function of LTPs and the formation of MCSs are regulated in cells remains poorly understood.
In the synthesis of sphingomyelin (SM) in the Golgi apparatus, the ceramide transport protein (CERT), a typical LTP, delivers the precursor ceramide synthesized in the ER to the Golgi apparatus, where ceramide is converted to SM [
3]. CERT contains a steroidogenic acute regulatory protein-related lipid transfer (START) domain, which catalyzes the inter-membrane transfer of ceramide as its lipid-transfer domain. In addition, to execute rapid and accurate ceramide transport at the ER–Golgi contact sites, CERT possesses the pleckstrin homology (PH) domain that preferentially recognizes phosphatidylinositol 4-monophosphate [PtdIns(4)P], which is the predominant phosphoinositide in the Golgi membranes, and two phenylalanines in an acidic tract (FFAT) motif that associates with VAMP-associated protein (VAP), an ER-resident protein [
4]. Of note, de novo synthesis of SM marginally occurs even in the absence of CERT [
3,
5,
6], suggesting the existence of a CERT-independent pathway for ER-to-Golgi transport of ceramide, which remains poorly characterized.
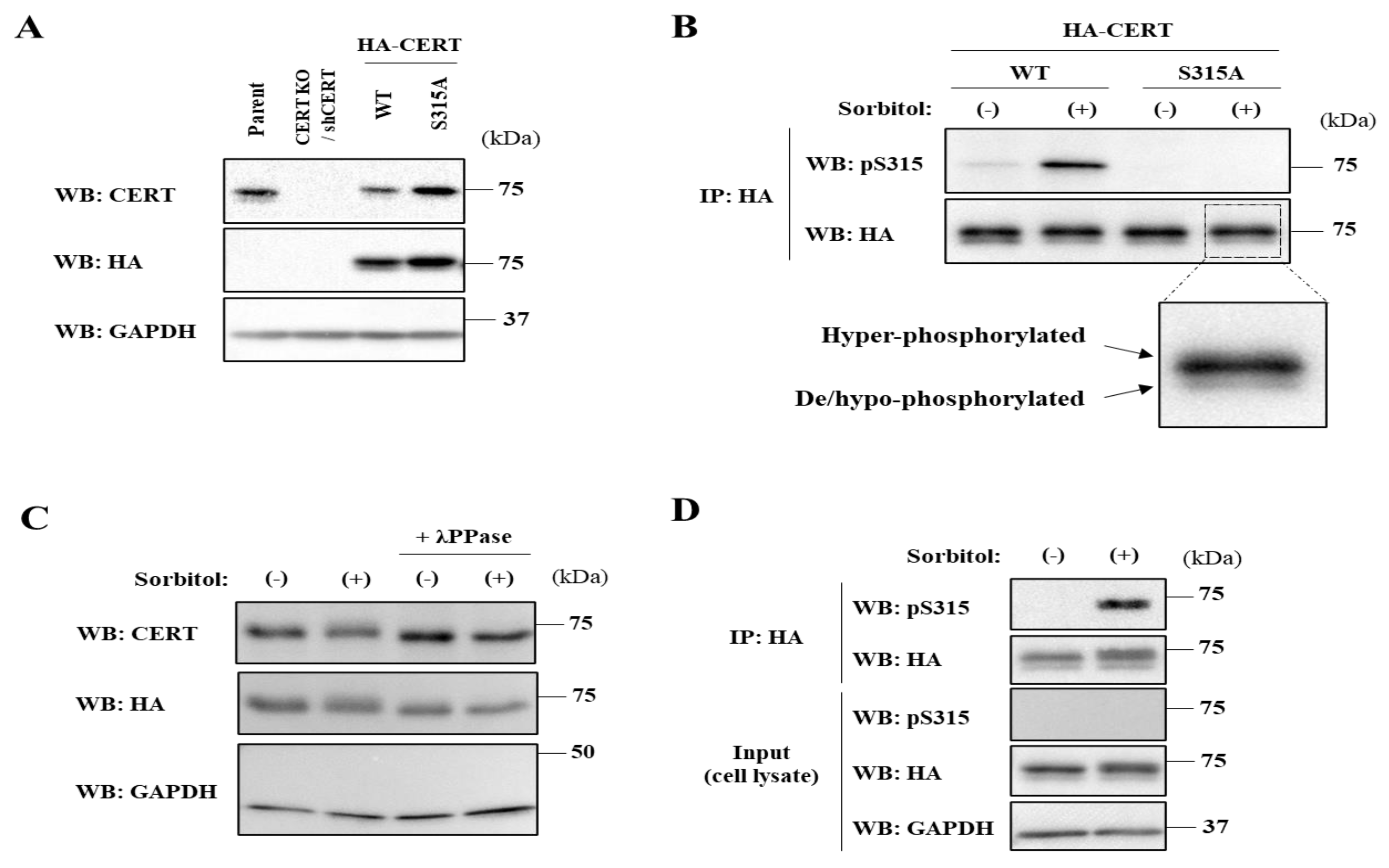
The function of CERT is regulated in at least two distinct phosphorylation-dependent events. One of the phosphorylation sites is a serine-repeat motif (SRM), which resides downstream of the PH domain. Once the first serine (S132) in the SRM is phosphorylated by protein kinase D [
7], the subsequent serine/threonine residues are sequentially phosphorylated by casein kinase 1ɤ [
8]. When the SRM receives multiple phosphorylations, the activities of the PH domain, FFAT motif, and START domain are simultaneously repressed, and the function of CERT is consequently inactivated [
9,
10]. The S132A mutant, which mimics the dephosphorylated SRM form of CERT, exhibits a constitutively active feature [
7,
9]. Another phosphorylation site in CERT is serine 315 (S315), which resides upstream of the FFAT motif. The phosphorylation of S315 results in an increase in the FFAT motif-dependent affinity of CERT for VAP, enhancing the ER-to-Golgi ceramide trafficking function of CERT [
10], although it remains unclear whether CERT is capable of binding to VAP anywhere in the ER or at the ER–Golgi MCSs. When de novo synthesis of SM is pharmacologically blocked, the SRM of CERT is dephosphorylated while S315 is phosphorylated [
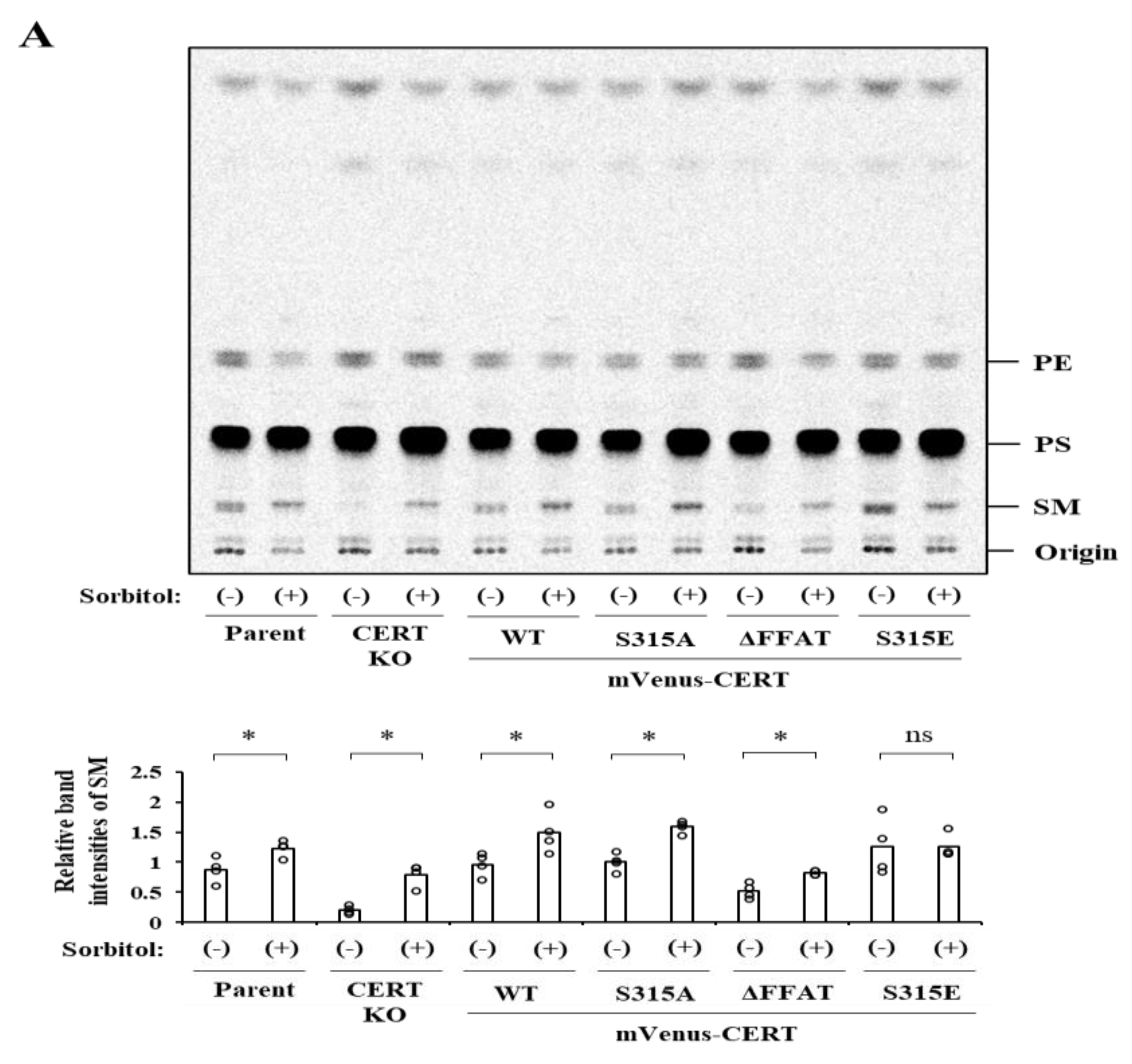
10], providing evidence that the two events of the SRM dephosphorylation and S315 phosphorylation are coordinated to fully activate CERT when the cells require SM. However, it remains unknown whether these two events are always coordinated.
In the present study, we found that hyperosmotic stress induces the phosphorylation of CERT S315 residue while the phosphorylation state of SRM remains multiply phosphorylated, which indicates that the two phosphorylation events can occur independently. Under hyperosmotic conditions, the interaction of CERT with VAP-A is enhanced in a CERT S315 phosphorylation-dependent manner, and this increased binding occurs throughout the ER rather than at the ER –Golgi MCSs. In addition, we unexpectedly found that de novo synthesis of SM with very-long acyl chains increases in a CERT-independent manner in hyperosmotic stressed cells, providing a feature of a CERT-independent ceramide transport pathway for de novo synthesis of SM.
3. Discussion
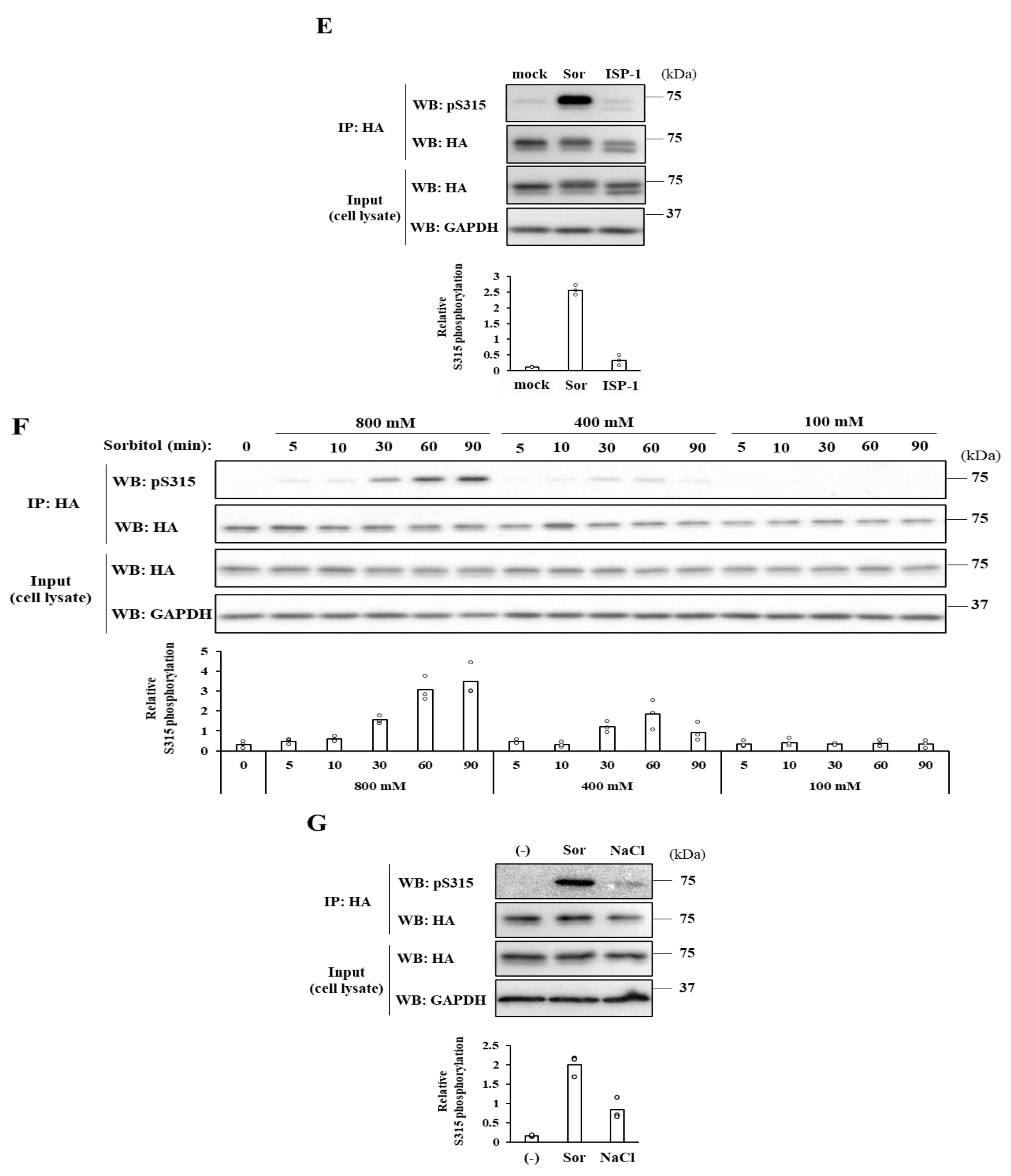
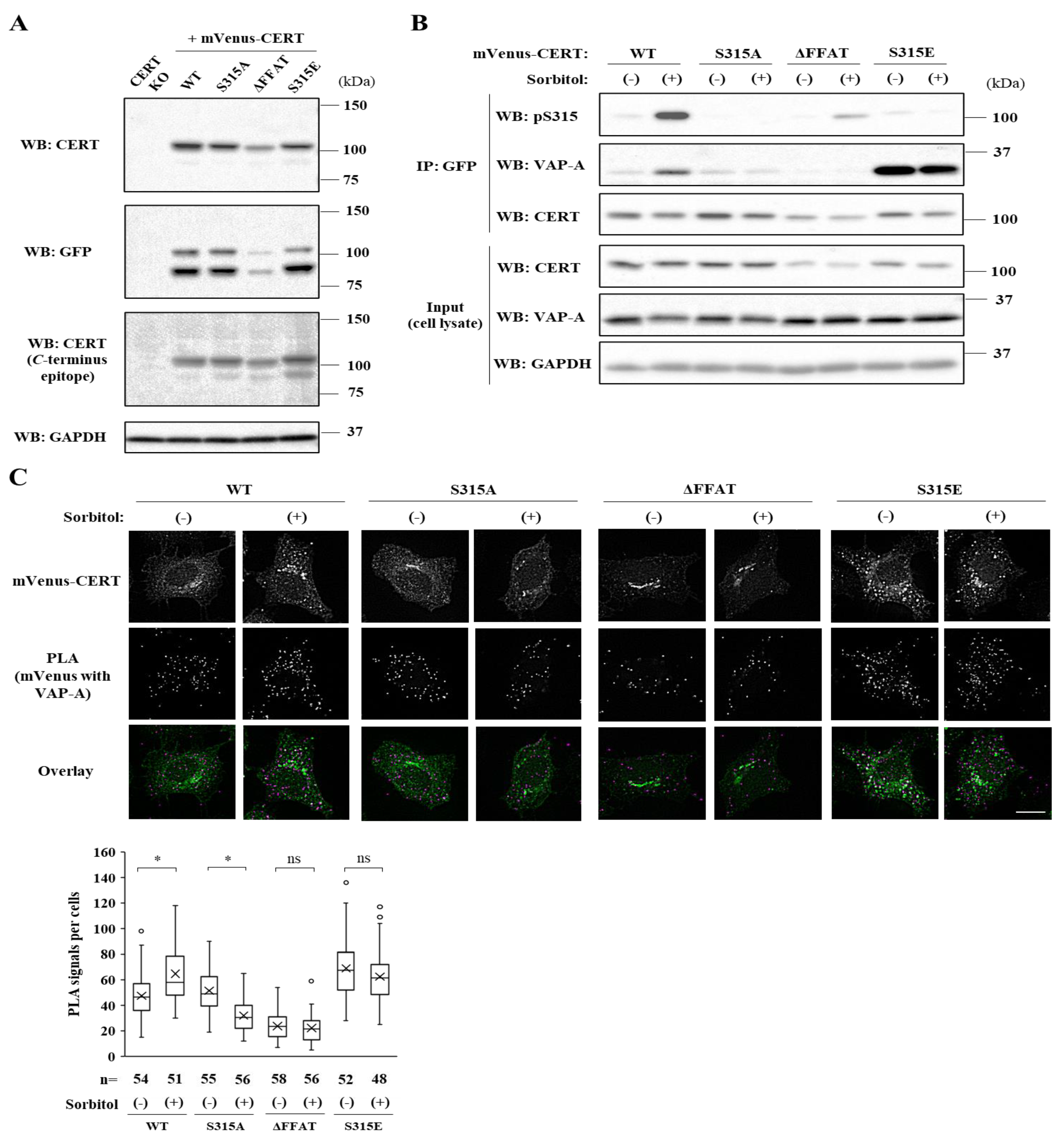
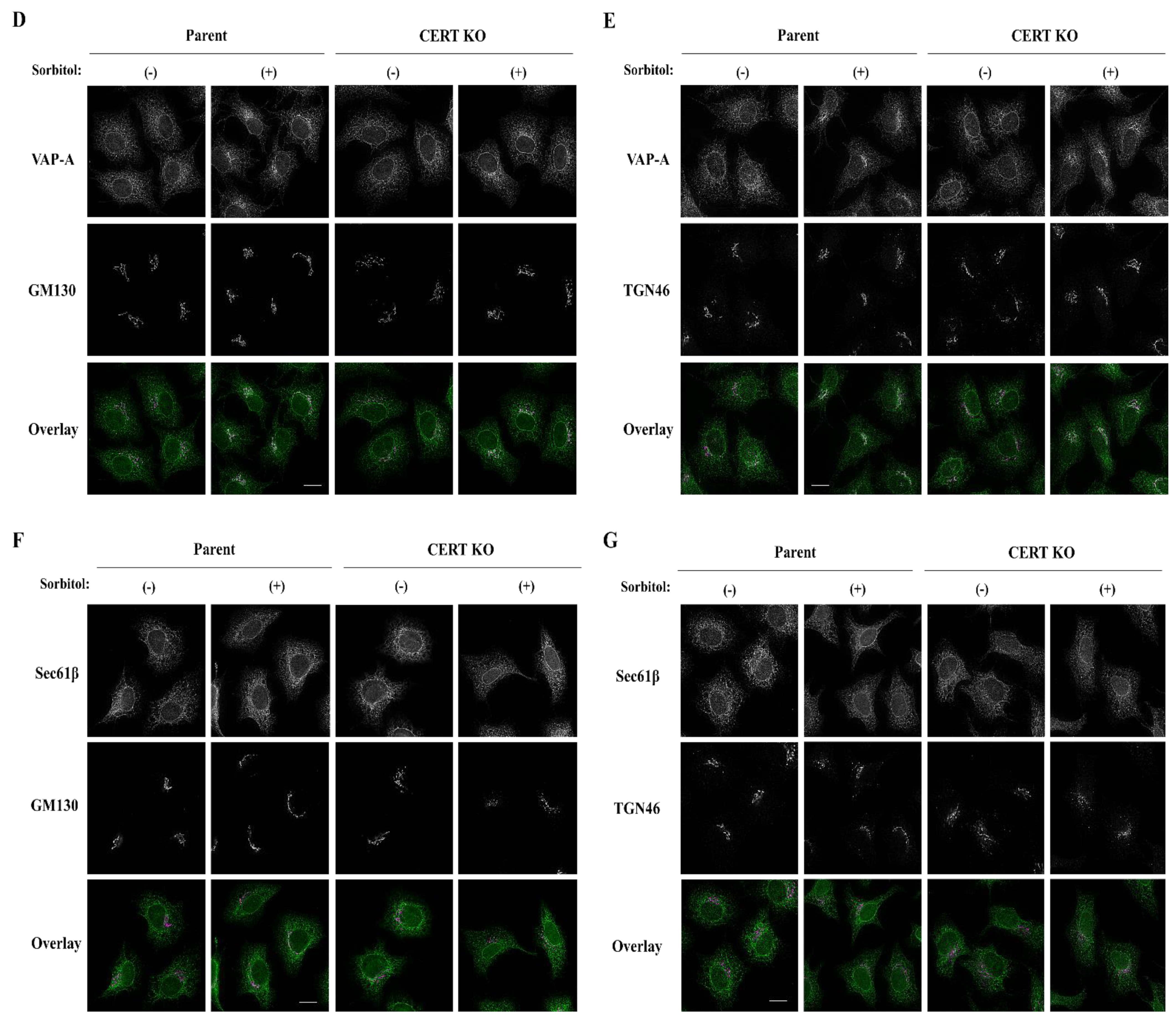
Both the SRM dephosphorylation and S315 phosphorylation occur to activate CERT fully [
9,
10]. Nevertheless, it remained unknown whether these two phosphorylation events are always induced simultaneously. In this study, we serendipitously found that hyperosmotic stress evokes CERT S315 phosphorylation without affecting the SRM-phosphorylation state. This demonstrates that the two phosphorylation events can be separated depending on the types of stimuli. Under hyperosmotic conditions, the binding of CERT with VAP-A is enhanced in an S315 phosphorylation- and FFAT motif-dependent manner, and this increased binding occurs widely on the ER (
Figure 6A), indicating that CERT has the potential to bind VAP anywhere on the ER and that it is not restricted to the ER–Golgi MCSs. When the PH domain of ER-scattering CERT meets PtdIns(4)P of
trans Golgi regions, which appose in close proximity to subregions of smooth ER [
20], CERT might be trapped at the ER–Golgi MCSs.
Although hyperosmotic stress invokes an increase in de novo SM synthesis in HeLa cells, this increase occurs independently of CERT (
Figure 4). Thus, the hyperosmotic stress-induced enhancement of CERT binding to the ER is unlikely to enhance CERT’s ER–Golgi ceramide trafficking function. We speculate that the enhanced binding of CERT to the ER is somehow beneficial for hyperosmotic stressed cells. Previous studies demonstrated that aberrant trafficking of ceramide to mitochondria causes cell death [
21,
22]. Cells undergo shrinkage under hyperosmotic conditions, which might generate otherwise absent emergent physical contacts between the ER and other organelles.
Moreover, hyperosmotic stress impinges on various types of membrane vesicle trafficking, such as the ER-to-Golgi transports or endosomal pathways [
23,
24]. These trafficking defects may also disrupt cellular homeostasis, including flows of sphingolipids. Therefore, it would be conceivable that trapping CERT on the ER prevents CERT from mis-sorting ceramide to non-Golgi organelles when abnormal organelle MCSs are generated or lipid metabolic enzymes are not appropriately sorted.
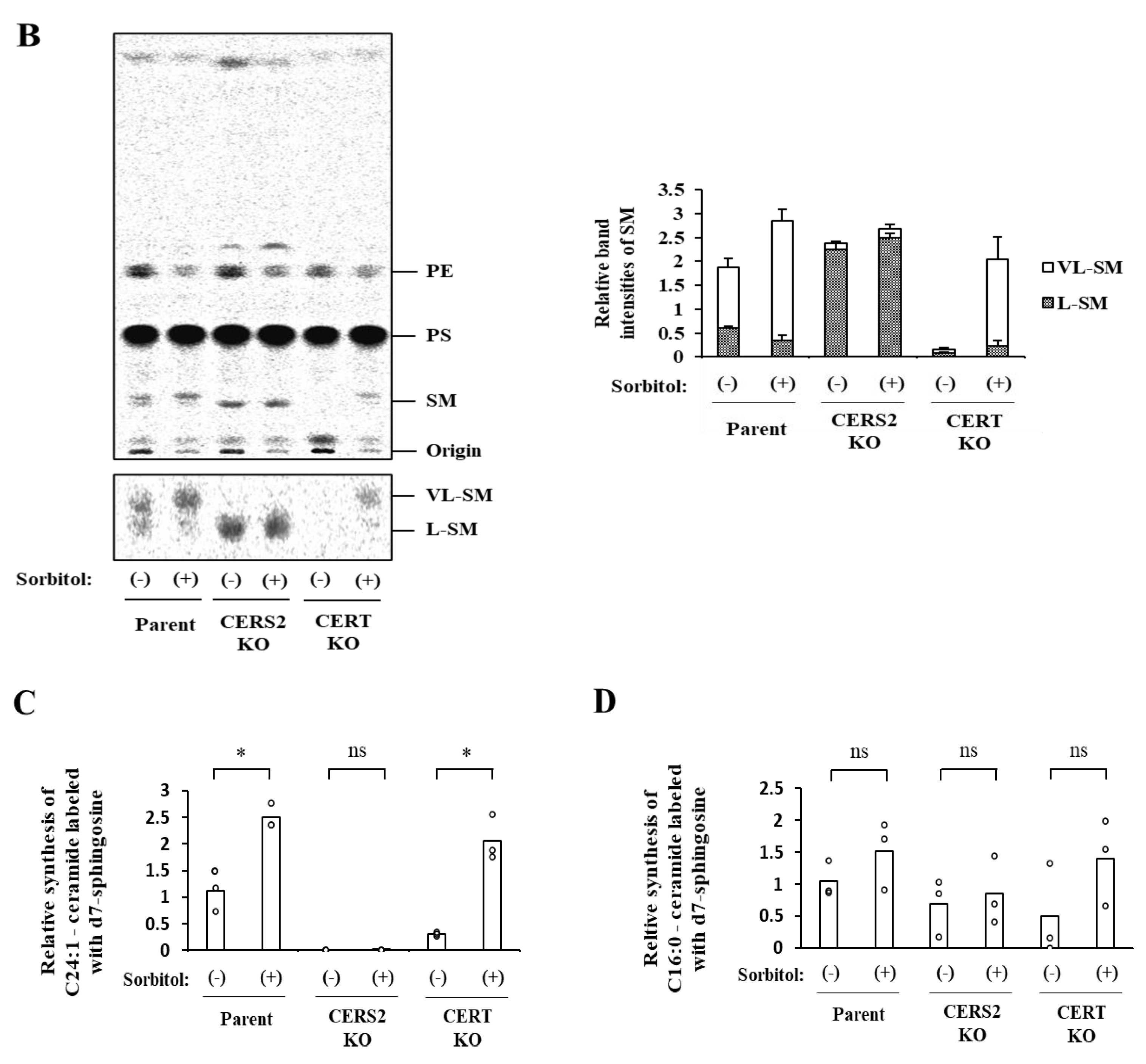
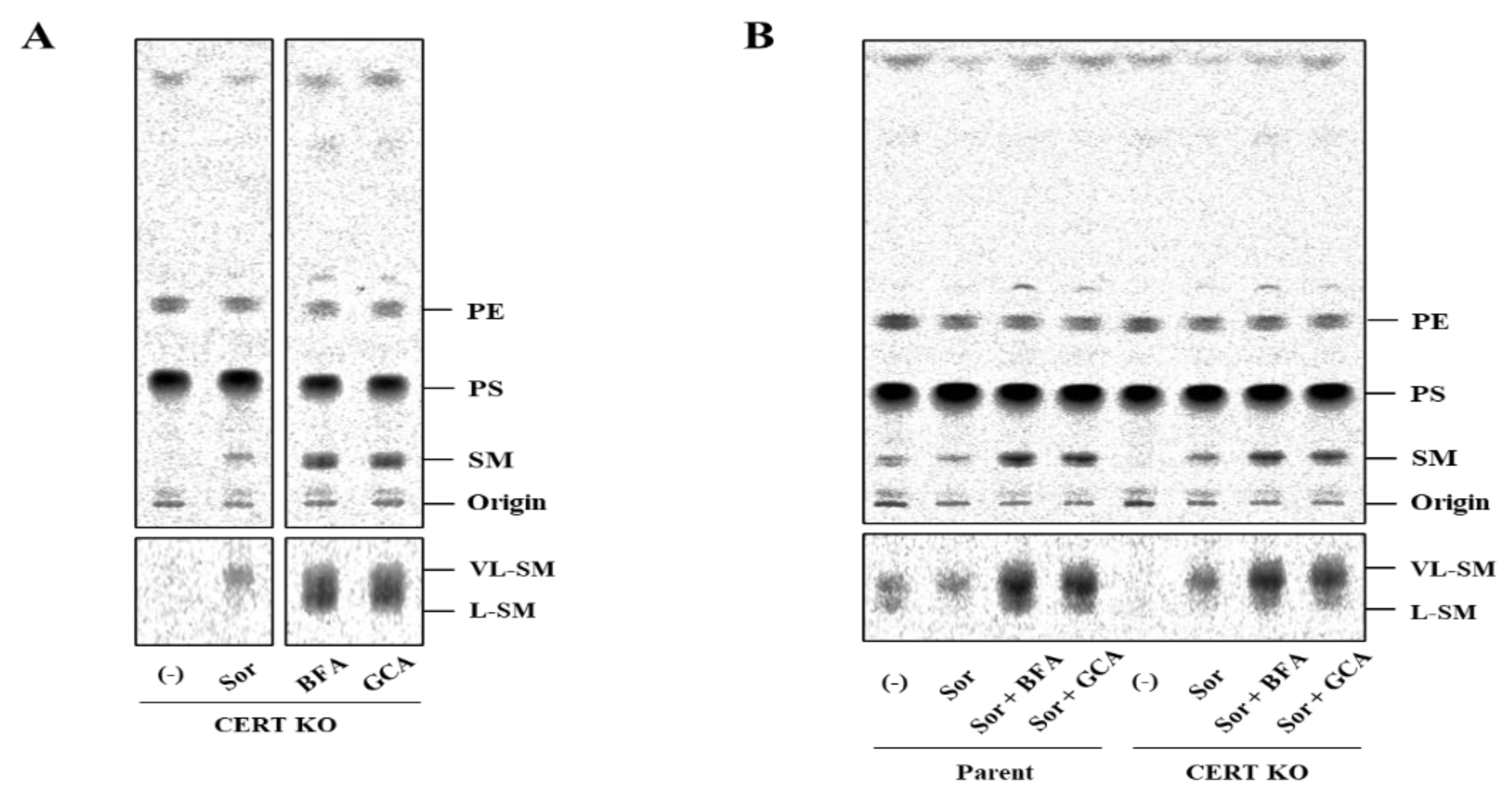
In addition, we found that hyperosmotic stress induces preferential biosynthesis of VL-SM in a CERT-independent manner (
Figure 4A and
Figure 5A). The hyperosmotic stress-induced synthesis of VL-SM is not abrogated, even when the ER and Golgi are merged using pharmacological tools (
Figure 5B), which eliminated the possibility that the phenomenon is due to an upregulation of a CERT-independent pathway(s) for ER-to-Golgi transport of ceramide. CERS2 is the predominant ceramide synthase (also known as dihydrosphingoine-
N-acyltransferase) isoform to produce VL-ceramides (more precisely, VL-dihydroceramides) in HeLa cells [
6]. In CERS2 KO HeLa cells, L-SM, instead of VL-SM, is predominantly synthesized, and the level of L-SM synthesis is not affected by hyperosmotic stress (
Figure 4A). To explain these results together with those of a previous study [
6], we propose the model illustrated in
Figure 6B. In wild-type HeLa cells under isosmotic conditions,
l-ceramides are delivered from the ER to the distal Golgi regions preferentially via the CERT-dependent pathway, whereas VL-ceramides are presumably transported to the proximal or broad Golgi regions via a CERT-independent pathway(s), the entity of which remains elusive. Under hyperosmotic conditions, the synthesis of VL-ceramides is enhanced, and therefore, a greater amount of VL-ceramides is delivered to the Golgi via the CERT-independent pathway, thereby enhancing the synthesis of VL-SM. This model would account for that hyperosmotic stress inducing the synthesis of VL-SM even in the absence of CERT and also would explain that hyperosmotic stress does not enhance L-SM synthesis in the absence of CERS2. More studies will be needed to address unresolved questions—for example, how does hyperosmotic stress enhance the CERS2-dependent synthesis of VL-ceramides, and what is the entity of the CERT-independent pathway?
The osmolarity of the medium supplemented with 800 mM sorbitol was about 1100 mOsm. The renal inner medulla is exposed to such severe hyperosmotic environments [
25]. SM molecular species with different acyl-chain lengths were shown to be distributed among different subregions of the kidney [
26]. It could be possible that the hyperosmotic stress-induced phosphorylation of CERT S315 partly underlies the region-dependent distribution of various SM molecular species in the kidney.
Although the biological importance of VL-SM under osmotic stress has not yet been elucidated, a previous study showed that an increase in VL-sphingolipids in yeast cells may strengthen membrane integrity and may endow some tolerance to hyperosmotic stress [
27]. By analogy, the increased VL-SM synthesis in mammalian cells might be an acute response for adapting to hyperosmotic conditions. Considering that ceramides act as modulators of various proteins [
28], the shift from
l-ceramides to VL-ceramides might affect cellular signaling events. For example, C16-ceramide, but not other ceramide species, stabilizes the p53 tumor suppressor, a key regulator in various fundamental cellular events such as the cell cycle, apoptosis, and survival in response to diverse stimuli [
29]. On the other hand, acute hyperosmotic stress induces caspase-mediated apoptosis [
30]. Thus, it is conceivable that a shift from
l-ceramides to VL-ceramides may destabilize p53 and consequently attenuate p53-mediated hyperosmotic stress-induced cell death. Further studies will be needed to address these possibilities.
In conclusion, hyperosmotic stress evokes CERT S315 phosphorylation without affecting the SRM-phosphorylation state, showing compelling evidence that the two phosphorylation events can be separated. Under hyperosmotic conditions, the binding of CERT with VAP-A is enhanced in an S315 phosphorylation- and FFAT motif-dependent manner, and this increased binding occurs widely on the ER. This reveals that CERT has the potential to bind VAP anywhere on the ER and that it is not restricted to the ER–Golgi MCSs.
4. Materials and Methods
4.1. HeLa Cell Lines and Cell Culture
The HeLa-mCAT#8 cell line that stably expresses the mouse ecotropic retroviral receptor mCAT-1 was used as the HeLa WT cell line, and its
CERT1-disrupted mutant cell line was used as the HeLa CERT KO cell line [
5] unless otherwise noted. To avoid unwanted overexpression of CERT cDNAs, the HeLa CERT KO/shCERT cell line was used. The HeLa CERT KO/shCERT cell line was established as described in a separate publication (Goto et al., submitted). In brief, the CERT KO cells were transfected using a retroviral vector containing a short hairpin RNA against
CERT1 RNA to interfere with the expression of
CERT1 mRNA. Then, a stable clone was isolated and used as the parental HeLa CERT KO/shCERT cell line. The CERS2 KO cell line (#16) was established as described previously [
6]. HeLa cells were cultured in high glucose Dulbecco’s modified Eagle medium (D-MEM; 044-29765, Wako, Osaka, Japan) supplemented with 10% heat-inactivated fetal calf serum.
4.2. Plasmids
cDNA fragments encoding the full-length human CERT wild-type and its mutants were digested from previously described vectors [
4,
9] and subcloned into pMX-IB vectors containing the human influenza HA tag or the mVenus tag.
4.3. Retroviral Transfection and Establishment of Stable Cell Lines
Retroviral transfection of HeLa cells with various cDNA constructs was performed using the pMXs-IB-based retroviral vector as described previously [
5]. After selecting transfected cells with 7.5 µg/mL blasticidin (#029-18701, Wako), cell clones were purified by limited dilution. Cell cloning was not performed for blasticidin-selected cells expressing mVenus-tagged CERT; instead, a population of cells expressing the fluorescent protein in a limited range was selected by FACS. After propagation, the FACS-selected cells were used for analysis.
4.4. Western Blotting
Cells were seeded under subconfluent conditions and were lysed in the lysis buffer (50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1 mM EGTA, 100 mM NaCl, 50 mM NaF, 5 mM sodium pyrophosphate, 10 mM disodium-β-glycerophosphate, cOmplete™ Protease Inhibitor Cocktail (#11836153001, Roche, Basel, Switzerland), 1% Phosphatase Inhibitor Cocktail 2 (P5726, Sigma-Aldrich, Burlington, MA, USA), and 1% Phosphatase Inhibitor Cocktail 3 (P0044, Sigma-Aldrich)) containing 1% Triton X-100. After centrifugation (14,000× rpm for 10 min at 4 °C), the supernatants of the lysates were collected, and the protein concentrations were quantified with the bicinchoninic acid (BCA) method (#23227, Thermo Scientific, Waltham, MA, USA). Equal volumes of the immunoprecipitated fraction or equal amounts of the total proteins in the input fraction were separated by electrophoresis on sodium dodecyl sulfate (SDS)/7.5% or 15% polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membrane (#1620177, Bio-Rad, Hercules, CA, USA). Precision Plus Protein™ All Blue Prestained Protein Standards (#1610373, Bio-Rad) were used as the molecular mass standards. After blocking, the membranes were immunoblotted using rabbit polyclonal anti-CERT antibody (ab72536, Abcam, Cambridge, UK), monoclonal anti-HA antibody (clone 3F10) conjugated with horseradish peroxidase (HRP) (#12013819001, Roche, Basel, Switzerland), mouse monoclonal anti-GAPDH antibody (clone 5A12, #016-25523, Wako), rabbit polyclonal anti-phospho-Serine315 CERT antibody generated by immunization [
10], rat monoclonal anti-GFP antibody (clone GF090R, #04404-26, Nacalai, Kyoto, Japan), rabbit polyclonal anti-VAP-A antibody (HPA009174, Sigma-Aldrich), and rabbit polyclonal anti-CERT
C-terminus antiserum generated by immunization [
9]. The designated proteins were visualized with the HRP-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) and SuperSignal™ West Femto Maximum Sensitivity Substrate (#34095, Thermo Scientific). The signals were captured using the LuminoGraph image analyzer (ATTO, Tokyo, Japan), and the images were processed with Fiji/ImageJ software (NIH). The relative levels of S315 phosphorylation were presented as the ratios of the band intensities normalized to those of the immunoprecipitated HA-CERT. The sum of the ratios in each condition was normalized to 3 (
Figure 1E,G), 16 (
Figure 1F), or 5 (
Figure 2A) (the number of conditions; arbitrary unit) in each experiment. The unprocessed images of the blots are shown in
Figure S1.
4.5. Protein Phosphatase Treatment
Cells were lysed with the lysis buffer without the phosphatase inhibitor cocktails, and the lysates (40 µL) were incubated with 800 units of λPPase (P0753, New England Biolabs, Ipswich, MA, USA) at 30 °C for 30 min.
4.6. Immunoprecipitation
Immunoprecipitation was performed as previously described [
10] with minor modifications. Briefly, cells were seeded in a 6-well plate under subconfluent conditions and were lysed in the lysis buffer containing 1% Triton X-100. After centrifugation, the supernatants were collected as the input fraction, and equal amounts of proteins were incubated with anti-HA antibody-conjugated agarose (A2095, Merck Millipore, Burlington, MA, USA) or anti-GFP antibody-conjugated agarose (#060830-05, Nacalai) for 1 h at 4 °C with gentle shaking. The resins were washed twice with the lysis buffer containing 0.1% Triton X-100 and were incubated with 1 × NuPAGE
® lithium dodecyl sulfate sample buffer with 50 mM dithiothreitol at 70 °C for 5 min. After centrifugation, the supernatants were collected and subjected to Western blotting as the immunoprecipitated fraction.
4.7. Immunocytochemistry
Cells were seeded on cover glasses (#12-545-80, Fisherbrand, Waltham, MA, USA) that were coated with collagen solution (TMTCC-050, TOYOBO, Tokyo, Japan) according to the manufacturer’s protocol. After 800 mM sorbitol treatment for 60 min at 37 °C, cells were fixed with Mildform 10N (#133-10311, Wako) for 15 min at room temperature. The cells were permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 10 min and incubated with a blocking solution containing 3% BSA in PBS for 30 min. The cells were incubated with the following primary antibodies: rat monoclonal anti-HA (#1867423001, Roche), rabbit polyclonal anti-VAP-A (HPA009174, Sigma-Aldrich), rabbit polyclonal anti-Sec61β (#07-205, Sigma-Aldrich), mouse monoclonal anti-GM130 (#610823, BD Biosciences, San Jose, CA, USA), or sheep polyclonal anti-TGN46 (AHP500, Bio-Rad). Then, the cells were incubated with the following secondary antibodies: Alexa Fluor 488-conjugated goat anti-rabbit (A11034, Invitrogen, Waltham, MA, USA), Alexa Fluor 594-conjugated goat anti-mouse (A11037, Invitrogen), or Alexa Fluor 594-conjugated donkey anti-sheep (#713-586-147, Jackson ImmunoResearch). The cover glasses were mounted on a slide glass (S1214, MATSUNAMI) using Fluoromount reagent (K024, Diagnostic BioSystems, Pleasanton, CA, USA). The cells were visualized using a wide-field fluorescence microscope, BioZero (BZ-X710, Keyence, Tokyo, Japan), equipped with a Plan Apo V 60 × 1.40 oil immersion objective. Haze reduction function (condition 3), which applies a no-neighbor deconvolution algorithm to the captured image, was used to eliminate fluorescence blurring caused by scattered light and to capture clear images with high contrast. The acquired images were processed with Fiji/ImageJ software (NIH).
4.8. In situ Proximity Ligation Assay (PLA)
Cells were seeded on the cover glasses coated with collagen solution and processed as described in
Section 4.7. permeabilization step. PLA was carried out using Duolink In situ Detection Reagents Red (DUO92008, Sigma-Aldrich) according to the manufacturer’s protocol. The cells were incubated with rat monoclonal anti-GFP antibody (#04404-26, Nacalai) and rabbit polyclonal anti-VAP-A antibody, followed by PLA probe anti-rat PLUS and PLA probe anti-rabbit MINUS (DUO92005, Sigma-Aldrich). The PLA probe anti-rat PLUS was prepared using Duolink In situ Probemaker PLUS (DUO92009, Sigma-Aldrich) against donkey anti-rat antibody (#712-005-150, Jackson ImmunoResearch) according to the manufacturer’s protocol. The cover glasses were mounted on a slide glass using Duolink In situ Mounting Medium with DAPI (DUO82040, Sigma-Aldrich), and the cells were analyzed by a BioZero digital microscope (Haze reduction function: condition 3) equipped with a Plan Apo V 60 × 1.40 oil immersion objective. The fluorescence and bright images were processed by Fiji/ImageJ software to measure the number of spots of PLA signals per cells.
4.9. Metabolic Labeling of Lipids with Radioactive Serine
Metabolic labeling of lipids with radioactive serine was performed as described previously [
5], except that cells were cultured in the normal culture medium overnight at 37 °C to subconfluence in a 12-well plate and then incubated in a serum-free DMEM containing 1.5 µCi of L-[U-
14C]-serine (MC-265, Moravek Inc., Brea, CA, USA) for 1 h under various osmotic conditions. The relative values of each lipid are presented as the ratios of the band intensities normalized to the amounts of proteins used in each condition. The sum of the ratios in each condition was normalized to 12 (
Figure 4A) or 6 (
Figure 4B) (the number of conditions; arbitrary unit) in each experiment. The unprocessed TLC images are shown in
Figure S1.
4.10. In Vitro Ceramide Synthesis Assay
HeLa cells were suspended in cell suspension buffer (50 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, 10% glycerol, 1 mM DTT, 1% Phosphatase Inhibitor Cocktail 2 (Sigma-Aldrich), 1% Phosphatase Inhibitor Cocktail 3 (Sigma-Aldrich), and cOmplete™ Protease Inhibitor Cocktail (Roche)) and then lysed by sonication. After removing the cell debris from the lysate by centrifugation (300×
g for 5 min at 4 °C), the supernatant fraction was centrifuged (100,000×
g for 30 min at 4 °C). The obtained participant, referred to as the membrane fraction, was suspended in cell suspension buffer and used as the enzyme source for the in vitro ceramide synthesis assay as follows. The membrane fraction (40–50 µg protein) was incubated with 5 µM deuterium-labeled (d7)-sphingosine (#860657P, Avanti Polar Lipids, Alabaster, AL, USA) and 25 µM C24:1- acyl CoA (#870725P, Avanti Polar Lipids) or C16:0-acyl CoA (P9716, Sigma-Aldrich) in 100 µL of the reaction buffer (the cell suspension buffer containing 2 mM MgCl2 and 0.1% digitonin) for 30 min at 37 °C. Following the addition of 1 nmol of C17 ceramide (#860517P, Avanti Polar Lipids) to the reaction mixture as an internal standard for LC–MS/MS, lipids were extracted using Bligh and Dyer’s method [
31], and the organic phase retrieved was evaporated and redissolved in 130 µL of methanol. Then, an aliquoted sample was subjected to an LC–MS system comprising a Prominence UFLC system (Shimadzu, Tokyo, Japan) coupled to a 3200 QTRAP System (SCIEX, Framingham, MA, USA), as previously described [
32]. The levels of declustering potential, entrance potential, and collision energy for deuterated ceramides were optimized for each target. The multiple-reaction monitoring (MRM) mode was used to measure the deuterated ceramides. The ion pairs of d7-d18:1/C16:0 ceramide and d7-d18:1/C24:1 ceramide were
m/z = 545.6–271.4 and 655.6–271.4, respectively. Each ion pair of the molecular species in MRM was monitored for 30 ms with a resolution of unit. The contents of individual ceramide were calculated by the peak area of analyte to the peak area of the internal standard. Data acquisition and analysis were performed using Analyst Software version 1.6. (SCIEX). The sum of the signals of each condition was normalized to 6 (the number of conditions; arbitrary unit) in each experiment.
4.11. Statistical Analysis
Statistical analyses were carried out using R software. The Student’s t-test was used for two-group comparisons, setting p < 0.05 as a statistical significance criterion.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








