
GZ17-6.02 Inhibits the Growth of EGFRvIII+ Glioblastoma

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
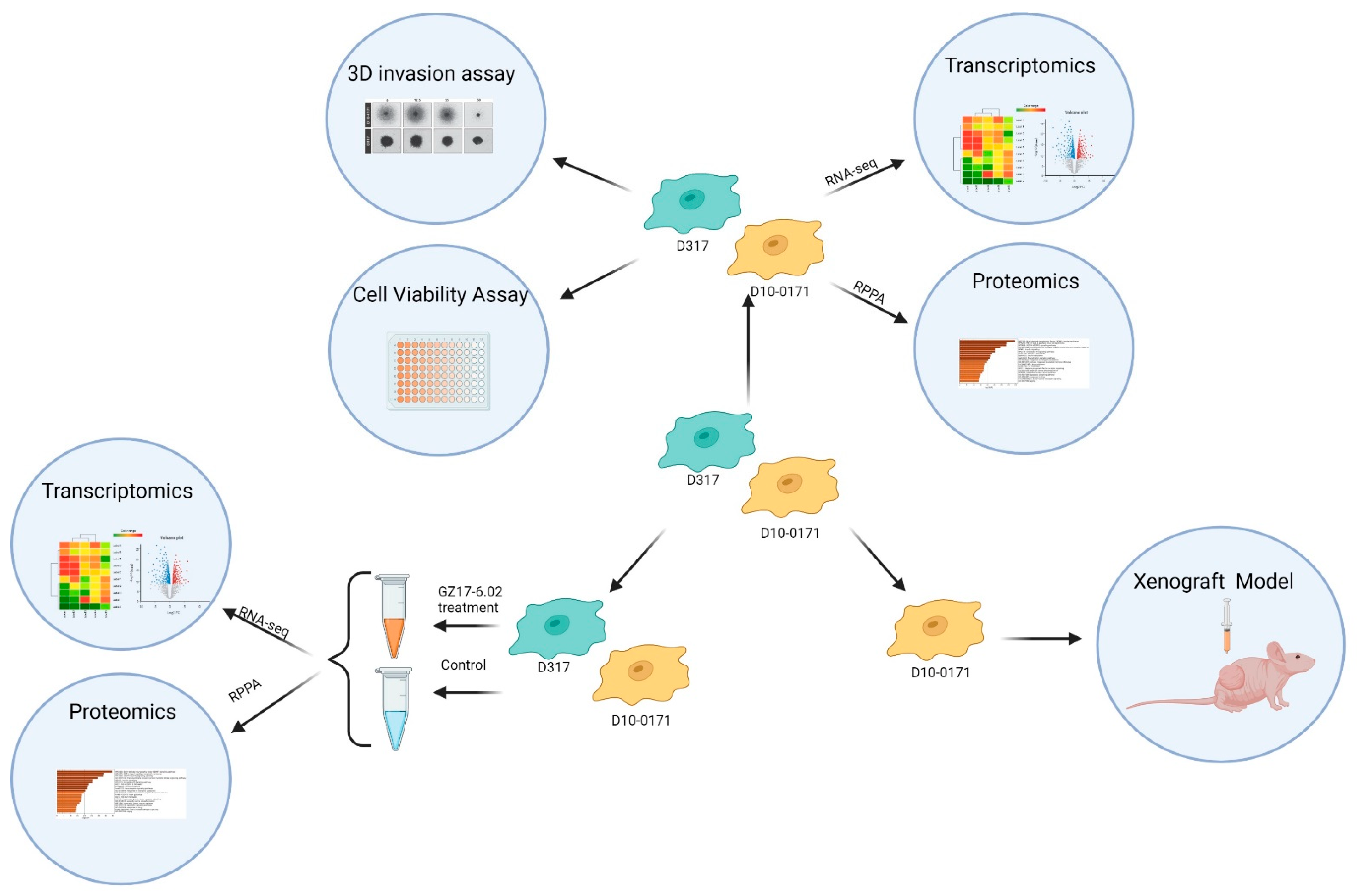
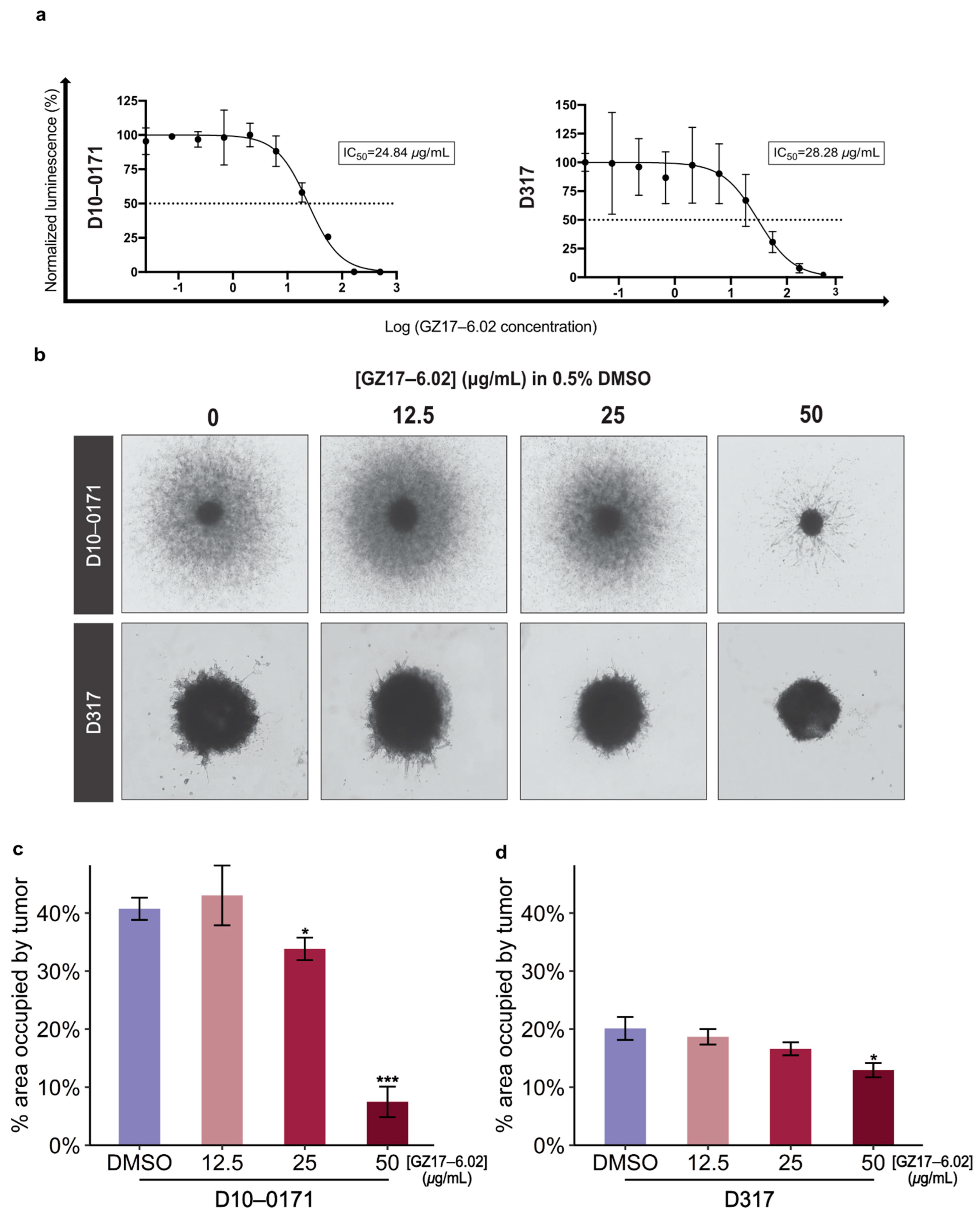
2.1. GZ17-6.02 Inhibits the Growth and Invasion of EGFRvIII+ GSCs
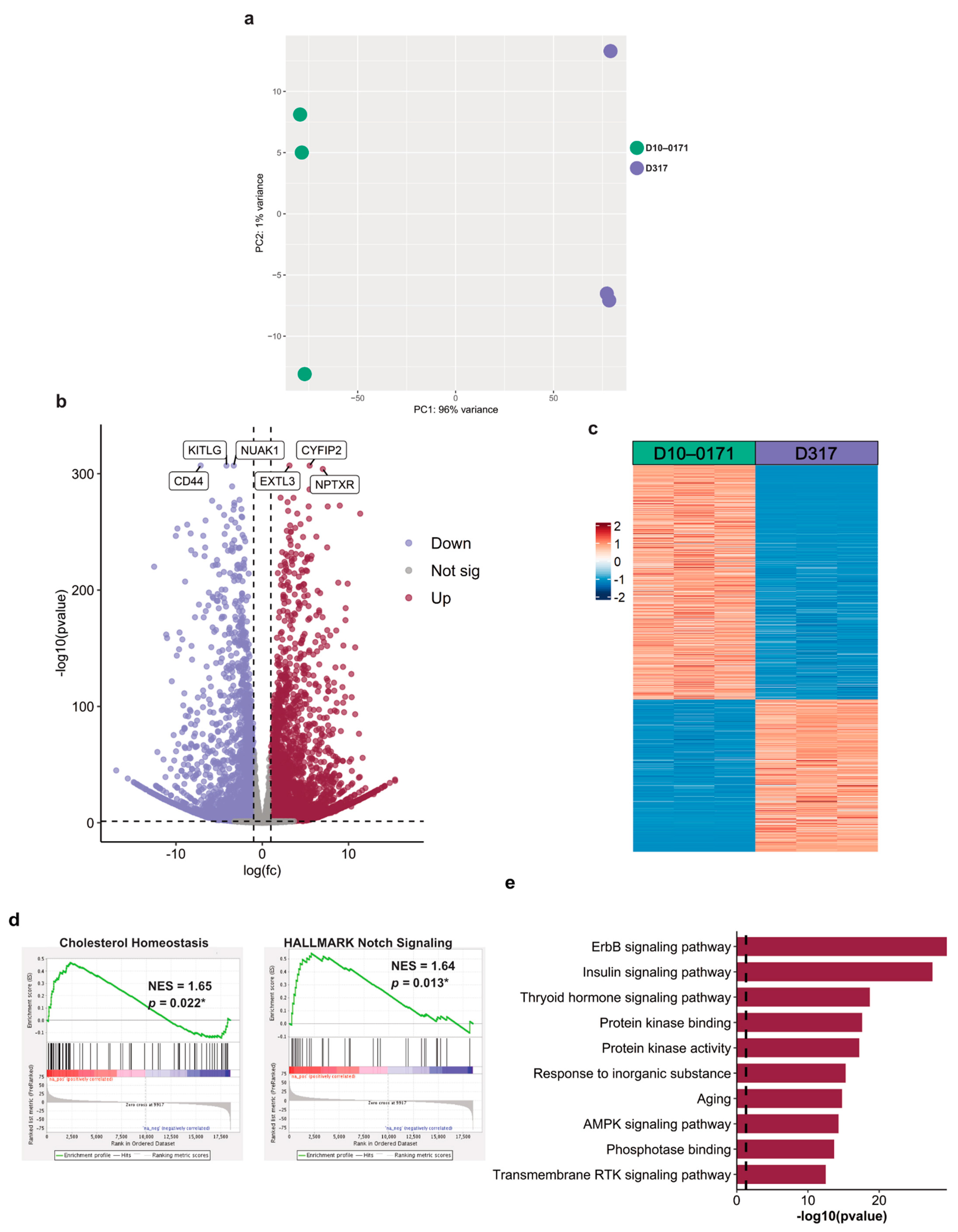
2.2. Molecular Differences between D10-0171 and D317
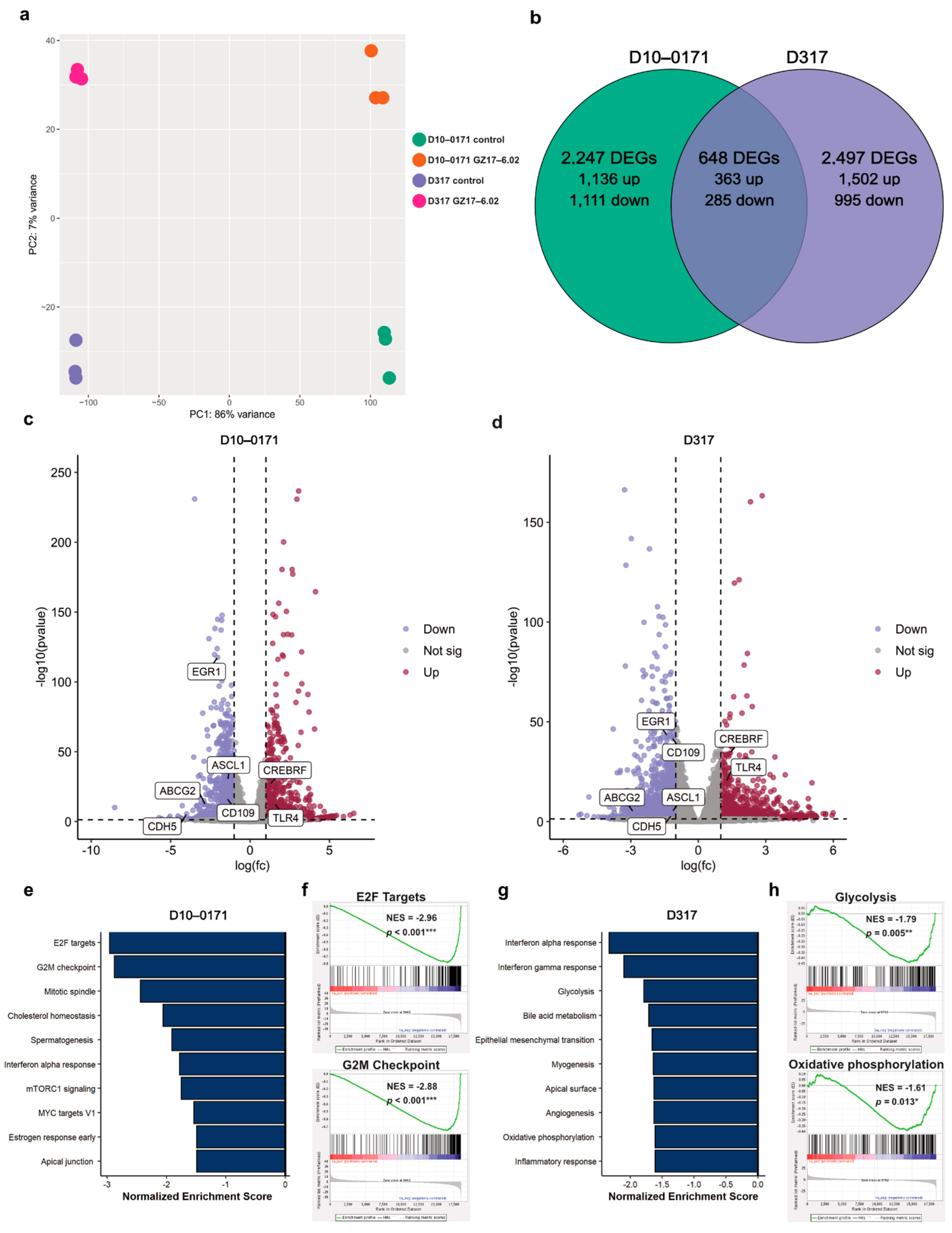
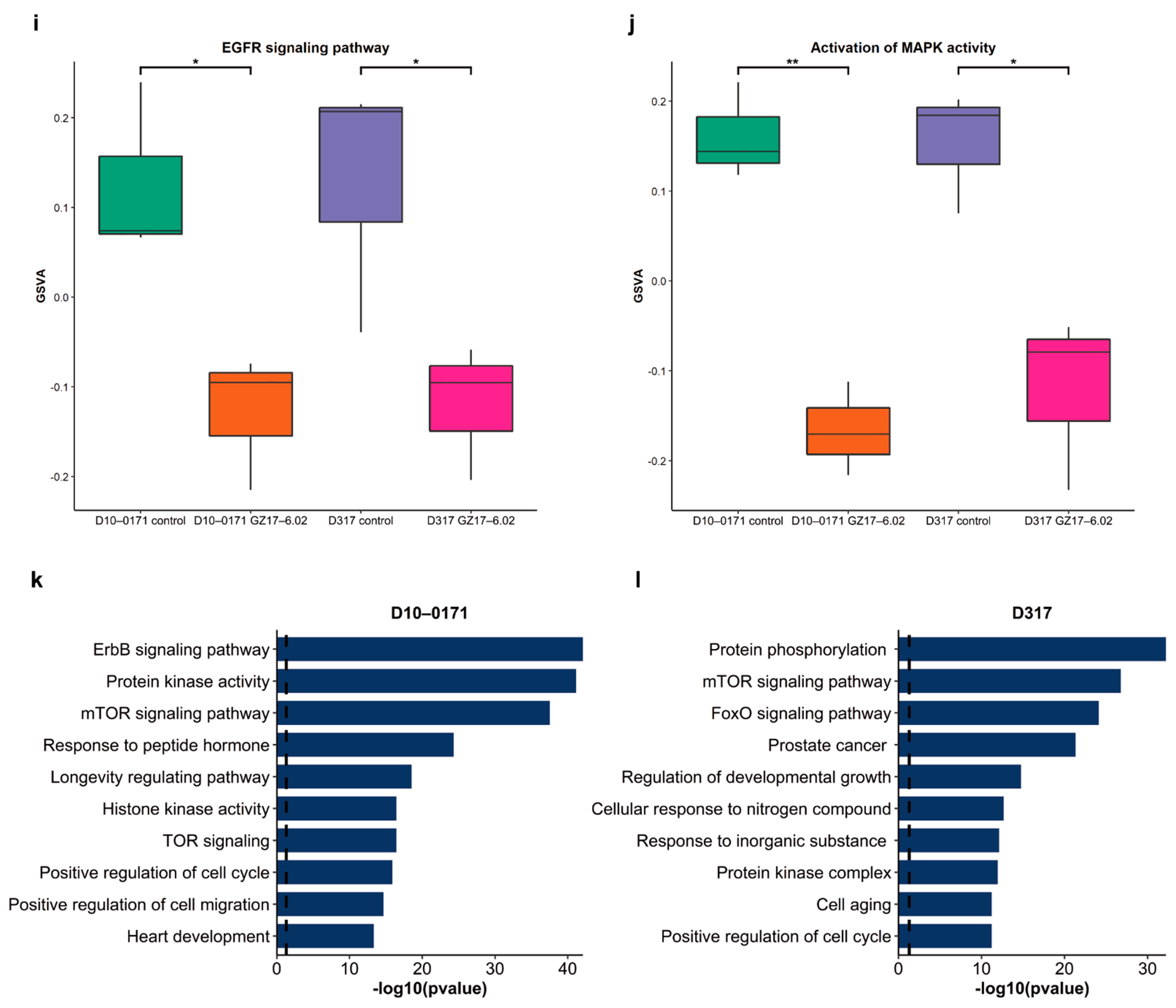
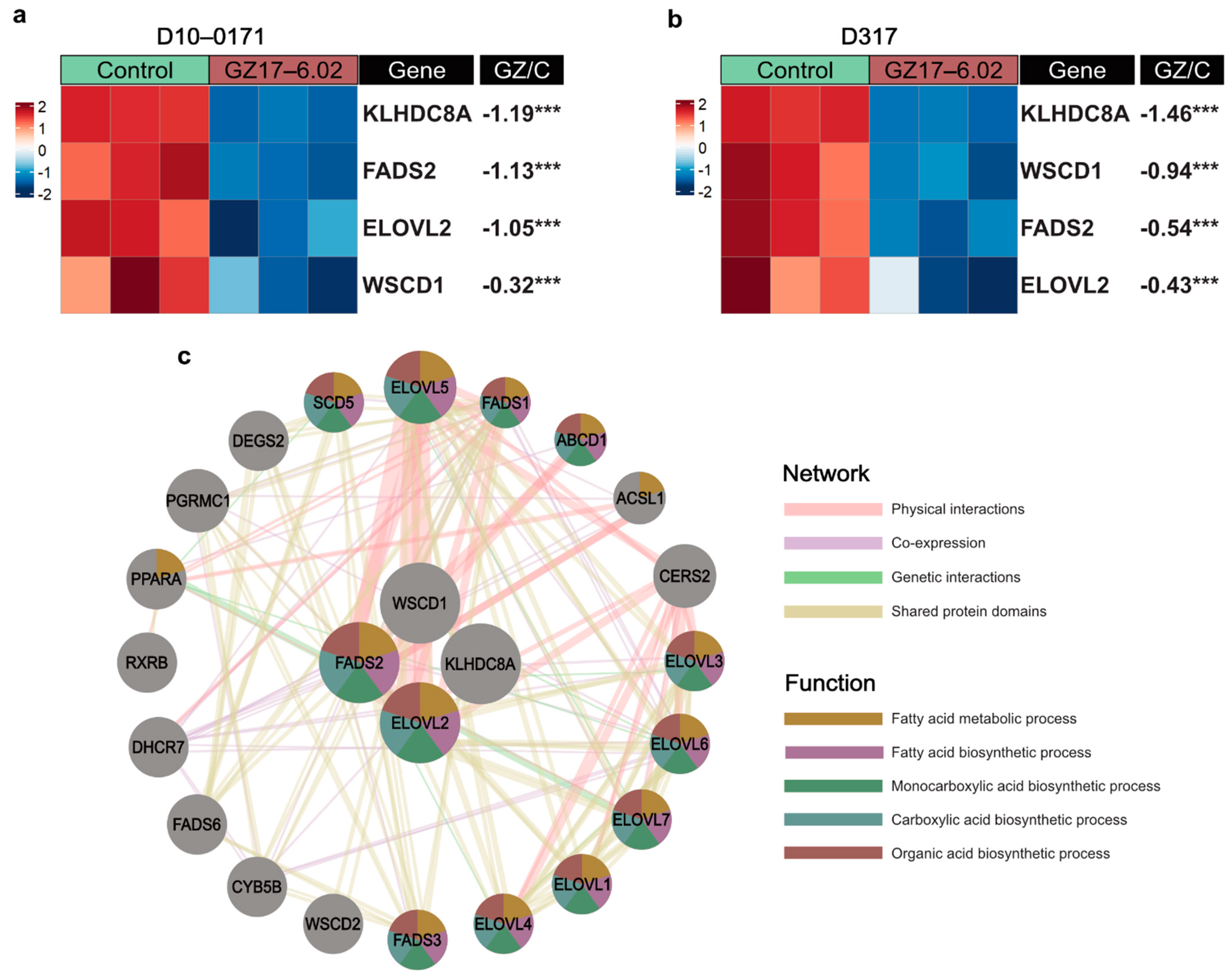
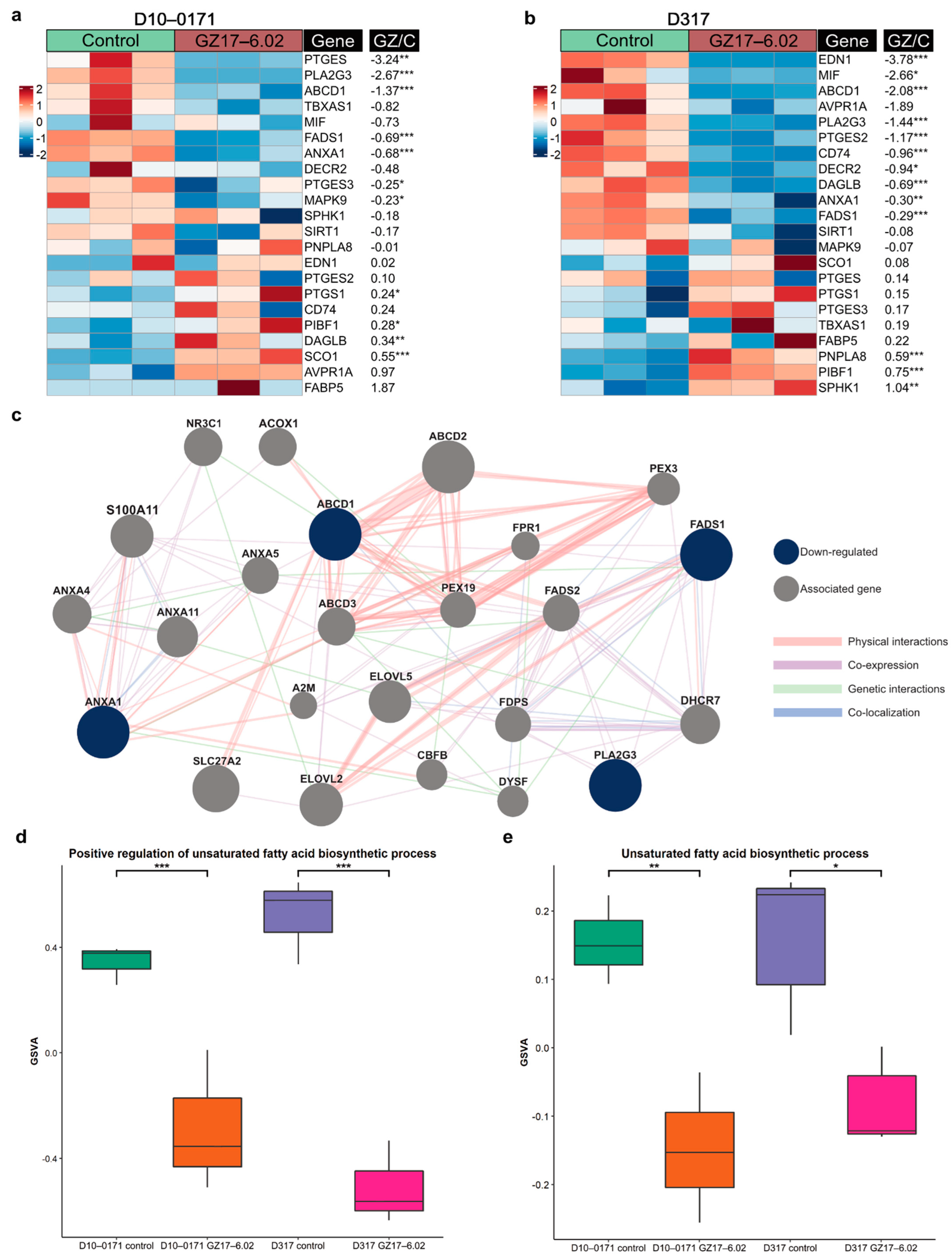
2.3. Transcriptomic and Proteomic Effects of GZ17-6.02 on EGFRvIII + GSCs
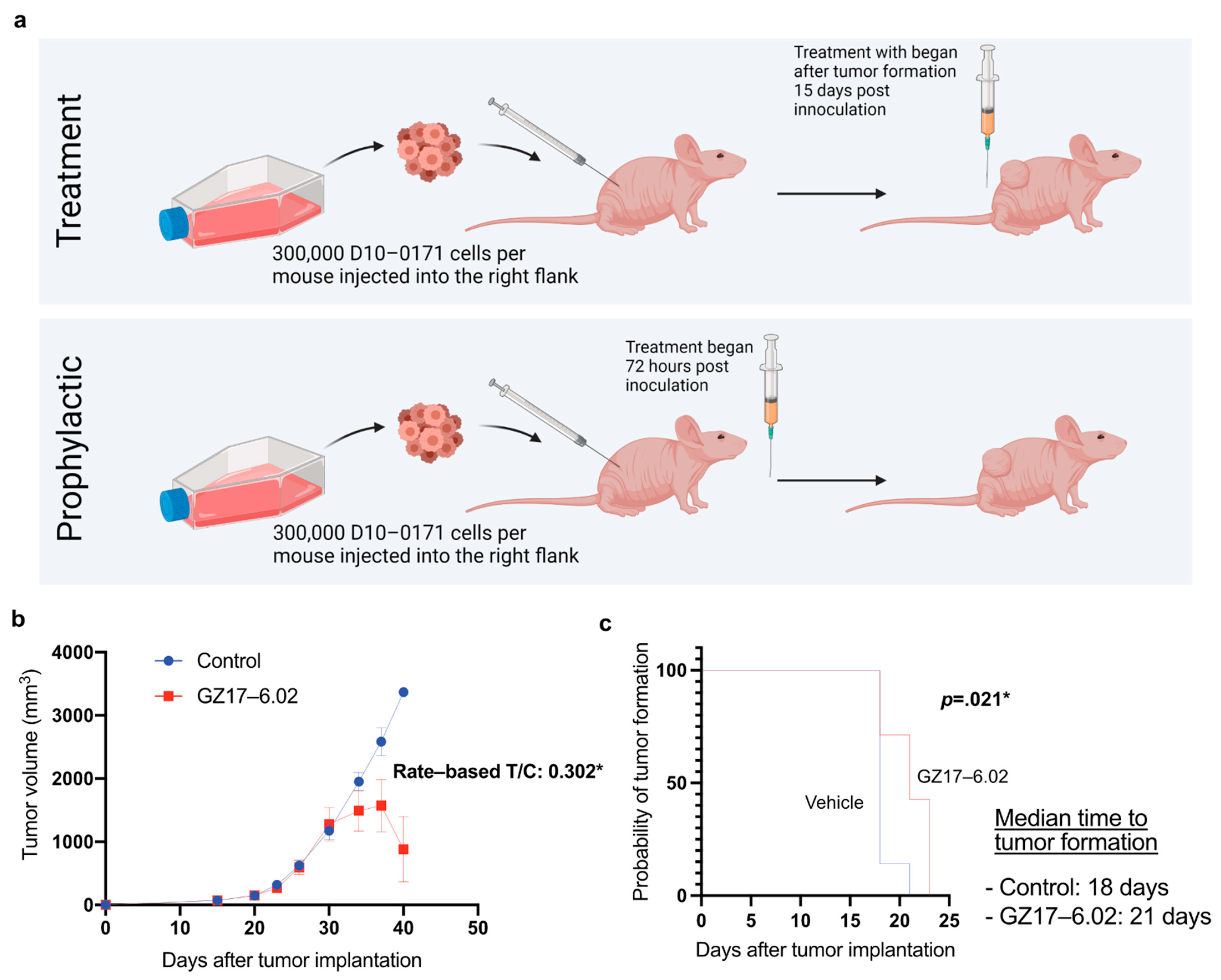
2.4. GZ17-6.02 Inhibits the Growth of D10-0171 GSCs in a Subcutaneous Tumor Model
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Cell Culture
4.3. Compounds and Reagents
4.4. Cell Viability Assays
4.5. Three-Dimensional Invasion Assays
4.6. Transcriptomic Analysis
4.7. Reverse Phase Protein Array (RPPA) Analysis
4.8. Subcutaneous Xenograft Tumor Model
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro. Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro-Oncol. 2018, 21, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Pottoo, F.H.; Javed, M.N.; Rahman, J.U.; Abu-Izneid, T.; Alam Khan, F. Targeted delivery of miRNA based therapeuticals in the clinical management of Glioblastoma Multiforme. Semin. Cancer Biol. 2021, 69, 391–398. [Google Scholar] [CrossRef]
- Gimple, R.C.; Kidwell, R.L.; Kim, L.J.Y.; Sun, T.; Gromovsky, A.D.; Wu, Q.; Wolf, M.; Lv, D.; Bhargava, S.; Jiang, L.; et al. Glioma Stem Cell-Specific Superenhancer Promotes Polyunsaturated Fatty-Acid Synthesis to Support EGFR Signaling. Cancer Discov. 2019, 9, 1248–1267. [Google Scholar] [CrossRef]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Kim, G.; Ko, Y.T. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch. Pharmacal Res. 2020, 43, 385–394. [Google Scholar] [CrossRef]
- Kwatra, M.M. A Rational Approach to Target the Epidermal Growth Factor Receptor in Glioblastoma. Curr. Cancer Drug Targets 2017, 17, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Chagoya, G.; Kwatra, S.G.; Nanni, C.W.; Roberts, C.M.; Phillips, S.M.; Nullmeyergh, S.; Gilmore, S.P.; Spasojevic, I.; Corcoran, D.L.; Young, C.C.; et al. Efficacy of osimertinib against EGFRvIII+ glioblastoma. Oncotarget 2020, 11, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.E.; Chagoya, G.; Kwatra, S.G.; Yen, T.; Keir, S.T.; Cooter, M.; Hoadley, K.A.; Rasheed, A.; Lipp, E.S.; McLendon, R.; et al. Proteomic profiling of patient-derived glioblastoma xenografts identifies a subset with activated EGFR: Implications for drug development. J. Neurochem. 2015, 133, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Booth, L.; Roberts, J.L.; West, C.; Von Hoff, D.; Dent, P. GZ17-6.02 initiates DNA damage causing autophagosome-dependent HDAC degradation resulting in enhanced anti-PD1 checkpoint inhibitory antibody efficacy. J. Cell. Physiol. 2020, 235, 8098–8113. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; West, C.; Von Hoff, D.; Dent, P. GZ17-6.02 and Doxorubicin Interact to Kill Sarcoma Cells via Autophagy and Death Receptor Signaling. Front. Oncol. 2020, 10, 1331. [Google Scholar] [CrossRef]
- Booth, L.; West, C.; Von Hoff, D.; Kirkwood, J.M.; Dent, P. GZ17-6.02 Interacts With [MEK1/2 and B-RAF Inhibitors] to Kill Melanoma Cells. Front. Oncol. 2021, 11, 656453. [Google Scholar] [CrossRef]
- Vishwakarma, V.; New, J.; Kumar, D.; Snyder, V.; Arnold, L.; Nissen, E.; Hu, Q.; Cheng, N.; Miller, D.; Thomas, A.R.; et al. Potent Antitumor Effects of a Combination of Three Nutraceutical Compounds. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Nozawa, T.; Okada, M.; Natsumeda, M.; Eda, T.; Abe, H.; Tsukamoto, Y.; Okamoto, K.; Oishi, M.; Takahashi, H.; Fujii, Y.; et al. EGFRvIII Is Expressed in Cellular Areas of Tumor in a Subset of Glioblastoma. Neurol. Med. -Chir. 2019, 59, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, B.; McEllin, B.; Camacho, C.V.; Tomimatsu, N.; Sirasanagandala, S.; Nannepaga, S.; Hatanpaa, K.J.; Mickey, B.; Madden, C.; Maher, E.; et al. EGFRvIII and DNA Double-Strand Break Repair: A Molecular Mechanism for Radioresistance in Glioblastoma. Cancer Res. 2009, 69, 4252–4259. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, P.A.; Wong, A.J.; Vogelstein, B.; Zalutsky, M.R.; Fuller, G.N.; Archer, G.E.; Friedman, H.S.; Kwatra, M.M.; Bigner, S.H.; Bigner, D.D. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc. Natl. Acad. Sci. USA 1990, 87, 4207–4211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.-I.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Ghosh, C.; Paul, S.; Dandawate, P.; Gunewardena, S.S.; Subramaniam, D.; West, C.; Anant, S.; Dhar, A. Super-enhancers: Novel target for pancreatic ductal adenocarcinoma. Oncotarget 2019, 10, 1554–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, L.; West, C.; Moore, R.P.; Von Hoff, D.; Dent, P. GZ17-6.02 and Pemetrexed Interact to Kill Osimertinib-Resistant NSCLC Cells That Express Mutant ERBB1 Proteins. Front. Oncol. 2021, 11, 711043. [Google Scholar] [CrossRef]
- Booth, L.; West, C.; Moore, R.P.; Von Hoff, D.; Dent, P. GZ17-6.02 and palbociclib interact to kill ER+ breast cancer cells. Oncotarget 2022, 13, 92–104. [Google Scholar] [CrossRef]
- Maiti, P.; Scott, J.; Sengupta, D.; Al-Gharaibeh, A.; Dunbar, G.L. Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/mTOR Pathway in Cultured Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 399. [Google Scholar] [CrossRef] [Green Version]
- Vallianou, N.G.; Evangelopoulos, A.; Schizas, N.; Kazazis, C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer Res. 2015, 35, 645–652. [Google Scholar]
- Chen, A.; Xu, J.; Johnson, A.C. Curcumin inhibits human colon cancer cell growth by suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene 2006, 25, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Zhen, L.; Fan, D.; Yi, X.; Cao, X.; Chen, D.; Wang, L. Curcumin inhibits oral squamous cell carcinoma proliferation and invasion via EGFR signaling pathways. Int. J. Clin. Exp. Pathol. 2014, 7, 6438–6446. [Google Scholar]
- Sun, X.-D.; Liu, X.-E.; Huang, D.-S. Curcumin induces apoptosis of triple-negative breast cancer cells by inhibition of EGFR expression. Mol. Med. Rep. 2012, 6, 1267–1270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, D.; Yu, S. Pharmacological effects of harmine and its derivatives: A review. Arch. Pharmacal Res. 2020, 43, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Pozo, N.; Zahonero, C.; Fernández, P.; Liñares, J.M.; Ayuso-Sacido, A.; Hagiwara, M.; Pérez, A.; Ricoy, J.R.; Hernández-Laín, A.; Sepúlveda, J.M.; et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Investig. 2013, 123, 2475–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Carvalho, A.C.; Girola, N.; de Figueiredo, C.R.; Machado, A.C.; de Medeiros, L.S.; Guadagnin, R.C.; Caseli, L.; Veiga, T.A.M. Understanding the cytotoxic effects of new isovanillin derivatives through phospholipid Langmuir monolayers. Bioorg. Chem. 2019, 83, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, G.; D’Auria, M.; Braca, A.; Mendez, J.; Castillo, A.; Morelli, I.; De Simone, A.F.; De Tommasi, N. Antioxidant and Free-Radical Scavenging Activity of Constituents of the Leaves of Tachigalia paniculata. J. Nat. Prod. 2002, 65, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.; Burgoyne, T.W.; Lee, A.; Stehno-Bittel, L.; Zaid, G.H. Arum Palaestinum with isovanillin, linolenic acid and β-sitosterol inhibits prostate cancer spheroids and reduces the growth rate of prostate tumors in mice. BMC Complement. Altern. Med. 2015, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Pirmoradi, L.; Seyfizadeh, N.; Ghavami, S.; Zeki, A.A.; Shojaei, S. Targeting cholesterol metabolism in glioblastoma: A new therapeutic approach in cancer therapy. J. Investig. Med. 2019, 67, 715–719. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef] [Green Version]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Multiforme Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [Green Version]
- Hather, G.; Liu, R.; Bandi, S.; Mettetal, J.; Manfredi, M.; Shyu, W.-C.; Donelan, J.; Chakravarty, A. Growth Rate Analysis and Efficient Experimental Design for Tumor Xenograft Studies. Cancer Inform. 2014, 13, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Song, B.-L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol metabolism: New functions and therapeutic approaches in cancer. Biochim. Et Biophys. Acta 2020, 1874, 188394. [Google Scholar] [CrossRef] [PubMed]
- Gorgisen, G.; Yaren, Z. Insulin receptor substrate 1 overexpression promotes survival of glioblastoma cells through AKT1 activation. Folia Neuropathol. 2020, 58, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tang, N.; Thompson, R.C.; Mobley, B.C.; Clark, S.W.; Sarkaria, J.N.; Wang, J. InsR/IGF1R Pathway Mediates Resistance to EGFR Inhibitors in Glioblastoma. Clin. Cancer Res. 2016, 22, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Huang, S. Role of mTOR Signaling in Tumor Cell Motility, Invasion and Metastasis. Curr. Protein Pept. Sci. 2011, 12, 30–42. [Google Scholar] [CrossRef]
- Wang, B.; Guo, H.; Yu, H.; Chen, Y.; Xu, H.; Zhao, G. The Role of the Transcription Factor EGR1 in Cancer. Front. Oncol. 2021, 11, 642547. [Google Scholar] [CrossRef]
- Du, K.; Wu, X.; Ji, X.; Liang, N.; Li, Z. Early growth response 1 promoted the invasion of glioblastoma multiforme by elevating HMGB1. J. Neurosurg. Sci. 2020. [Google Scholar] [CrossRef]
- Vue, T.Y.; Kollipara, R.K.; Borromeo, M.D.; Smith, T.; Mashimo, T.; Burns, D.K.; Bachoo, R.M.; Johnson, J.E. ASCL1 regulates neurodevelopmental transcription factors and cell cycle genes in brain tumors of glioma mouse models. Glia 2020, 68, 2613–2630. [Google Scholar] [CrossRef]
- Filppu, P.; Ramanathan, J.T.; Granberg, K.J.; Gucciardo, E.; Haapasalo, H.; Lehti, K.; Nykter, M.; Le Joncour, V.; Laakkonen, P. CD109-GP130 interaction drives glioblastoma stem cell plasticity and chemoresistance through STAT3 activity. JCI Insight 2021, 6, e141486. [Google Scholar] [CrossRef]
- Müller, P.; Gaber, S.A.A.; Zimmermann, W.; Wittig, R.; Stepp, H. ABCG2 influence on the efficiency of photodynamic therapy in glioblastoma cells. J. Photochem. Photobiol. B Biol. 2020, 210, 111963. [Google Scholar] [CrossRef] [PubMed]
- Wee, B.; Pietras, A.; Ozawa, T.; Bazzoli, E.; Podlaha, O.; Antczak, C.; Westermark, B.; Nelander, S.; Uhrbom, L.; Forsberg-Nilsson, K.; et al. ABCG2 regulates self-renewal and stem cell marker expression but not tumorigenicity or radiation resistance of glioma cells. Sci. Rep. 2016, 6, 25956. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.-G.; Xue, X.-Y.; Wang, L.; Zhang, X.; Yan, M.; Tu, Y.-Y.; Lin, W.; Jiang, X.-F.; Ren, H.-G.; Zhang, W.; et al. CDH5 is specifically activated in glioblastoma stemlike cells and contributes to vasculogenic mimicry induced by hypoxia. Neuro-Oncol. 2013, 15, 865–879. [Google Scholar] [CrossRef] [Green Version]
- Modarresi, F.; Faghihi, M.A.; Lopez-Toledano, M.A.; Fatemi, R.P.; Magistri, M.; Brothers, S.; Van Der Brug, M.P.; Wahlestedt, C. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 2012, 30, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Ghafouri-Fard, S.; Khoshbakht, T.; Taheri, M.; Ghanbari, M. A concise review on the role of BDNF-AS in human disorders. Biomed. Pharmacother. 2021, 142, 112051. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Ma, J.; Zheng, J.; Liu, X.; Liu, Y.; Ruan, X.; Shen, S.; Yang, C.; Wang, D.; Cai, H.; et al. PABPC1-induced stabilization of BDNF-AS inhibits malignant progression of glioblastoma cells through STAU1-mediated decay. Cell Death Dis. 2020, 11, 81. [Google Scholar] [CrossRef]
- Lv, X.; Gu, C.; Guo, S. Activation of BDNF-AS/ADAR/p53 positive feedback loop inhibits glioblastoma cell proliferation. Neurochem. Res. 2020, 45, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Zhang, J.; Guo, X.; Wang, J.; Li, J.; Gao, X.; Guo, X.; Li, T.; Xu, S.; Zhang, P.; et al. CREBRF is a potent tumor suppressor of glioblastoma by blocking hypoxia-induced autophagy via the CREB3/ATG5 pathway. Int. J. Oncol. 2016, 49, 519–528. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef]
- da Cruz, L.L.P.; de Souza, P.O.; Dal Prá, M.; Falchetti, M.; de Abreu, A.M.; Azambuja, J.H.; Bertoni, A.P.S.; Paz, A.H.R.; Araújo, A.B.; Visioli, F.; et al. TLR4 expression and functionality are downregulated in glioblastoma cells and in tumor-associated macrophages: A new mechanism of immune evasion? Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166155. [Google Scholar] [CrossRef]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irtenkauf, S.M.; Sobiechowski, S.; Hasselbach, L.A.; Nelson, K.K.; Transou, A.D.; Carlton, E.T.; Mikkelsen, T.; Decarvalho, A.C. Optimization of Glioblastoma Mouse Orthotopic Xenograft Models for Translational Research. Comp. Med. 2017, 67, 300–314. [Google Scholar] [PubMed]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assay for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersey, P.J.; Staines, D.M.; Lawson, D.; Kulesha, E.; Derwent, P.; Humphrey, J.C.; Hughes, D.S.T.; Keenan, S.; Kerhornou, A.; Koscielny, G.; et al. Ensembl Genomes: An integrative resource for genome-scale data from non-vertebrate species. Nucleic Acids Res. 2011, 40, D91–D97. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Bordeaux, Z.A.; McKeel, J.; Nanni, C.; Sutaria, N.; Braun, G.; Davis, C.; Miller, M.N.; Alphonse, M.P.; Kwatra, S.G.; et al. GZ17-6.02 Inhibits the Growth of EGFRvIII+ Glioblastoma. Int. J. Mol. Sci. 2022, 23, 4174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084174
Choi J, Bordeaux ZA, McKeel J, Nanni C, Sutaria N, Braun G, Davis C, Miller MN, Alphonse MP, Kwatra SG, et al. GZ17-6.02 Inhibits the Growth of EGFRvIII+ Glioblastoma. International Journal of Molecular Sciences. 2022; 23(8):4174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084174
Chicago/Turabian StyleChoi, Justin, Zachary A. Bordeaux, Jaimie McKeel, Cory Nanni, Nishadh Sutaria, Gabriella Braun, Cole Davis, Meghan N. Miller, Martin P. Alphonse, Shawn G. Kwatra, and et al. 2022. "GZ17-6.02 Inhibits the Growth of EGFRvIII+ Glioblastoma" International Journal of Molecular Sciences 23, no. 8: 4174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084174