
Differential Effects of Cytopathic Hypoxia on Human Retinal Endothelial Cellular Behavior: Implication for Ischemic Retinopathies

 ,
, 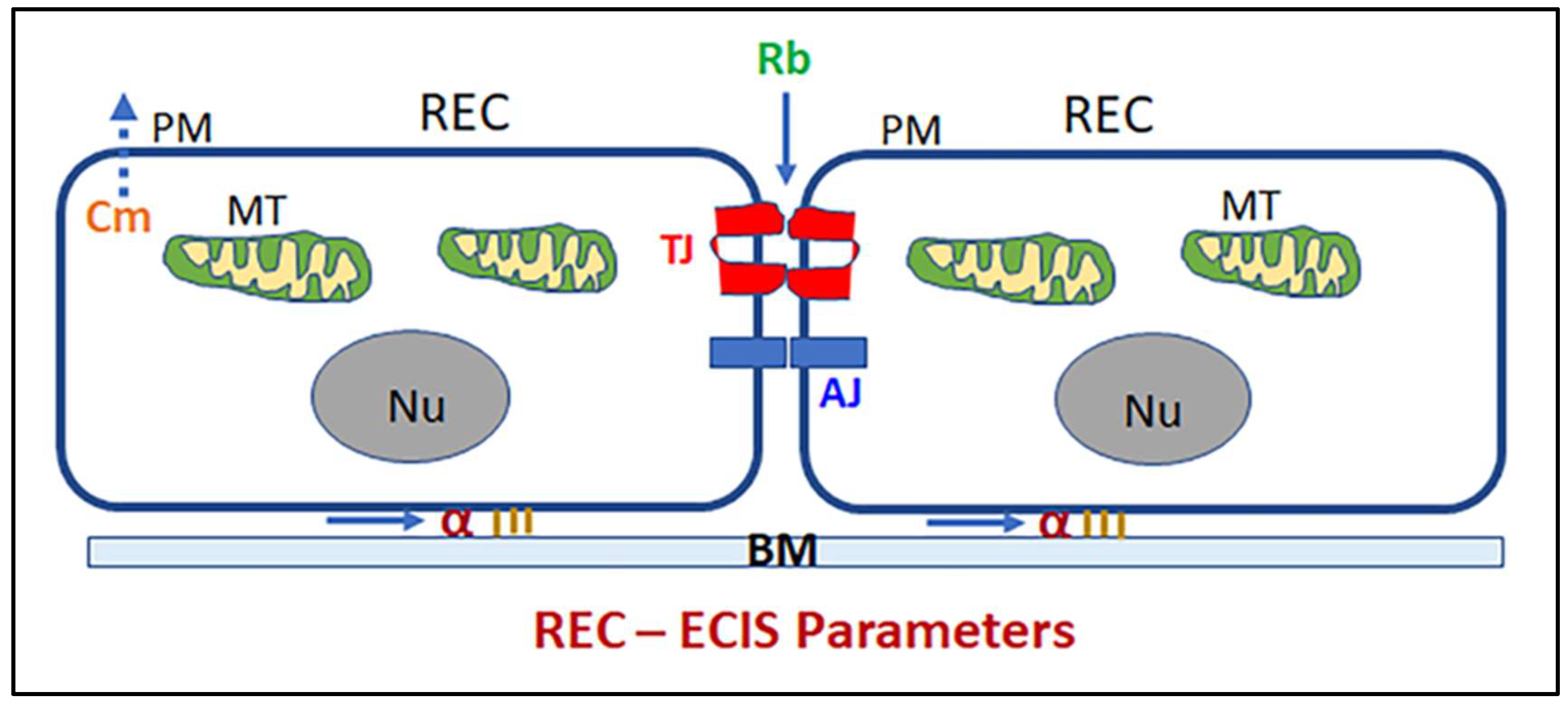{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
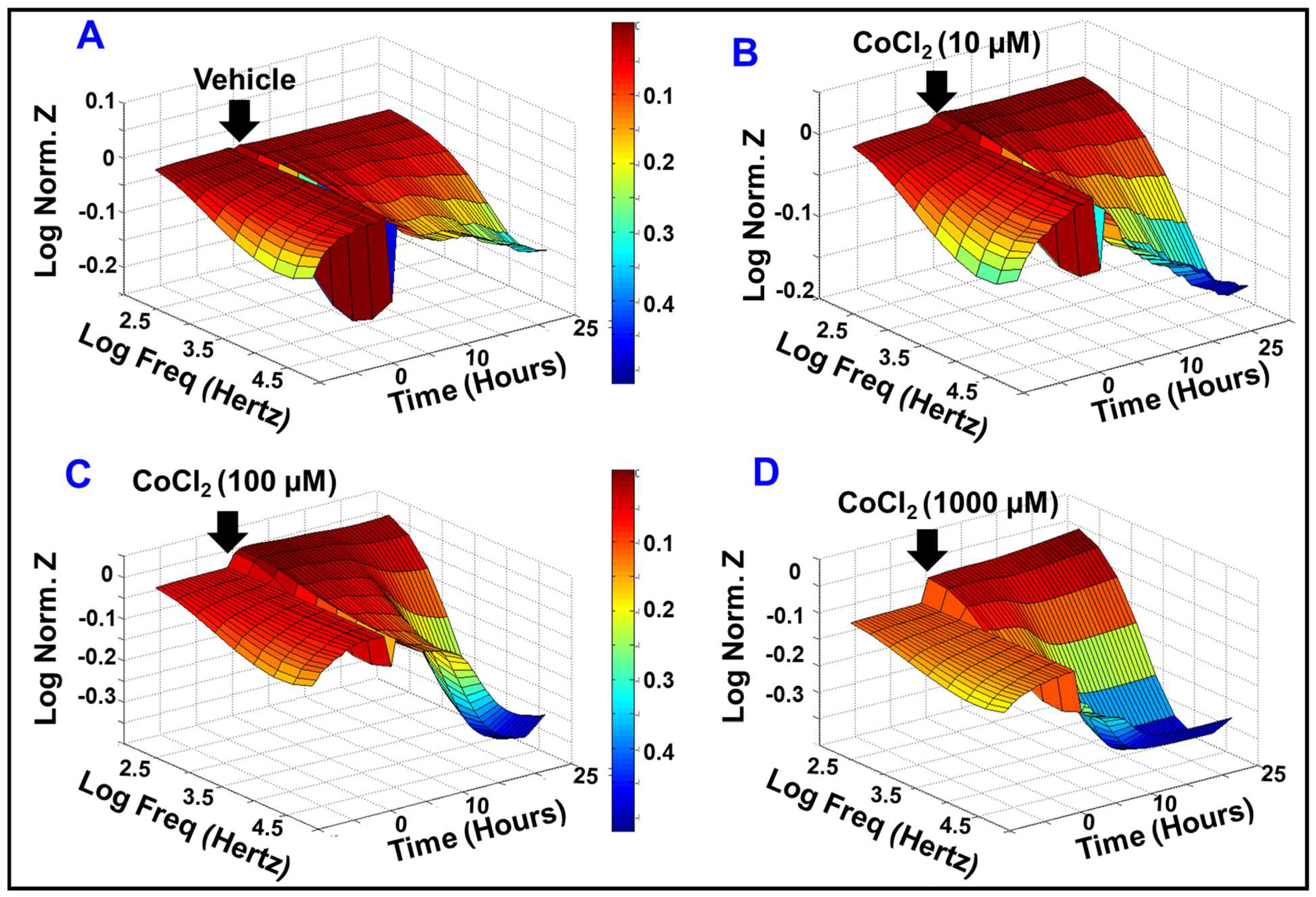
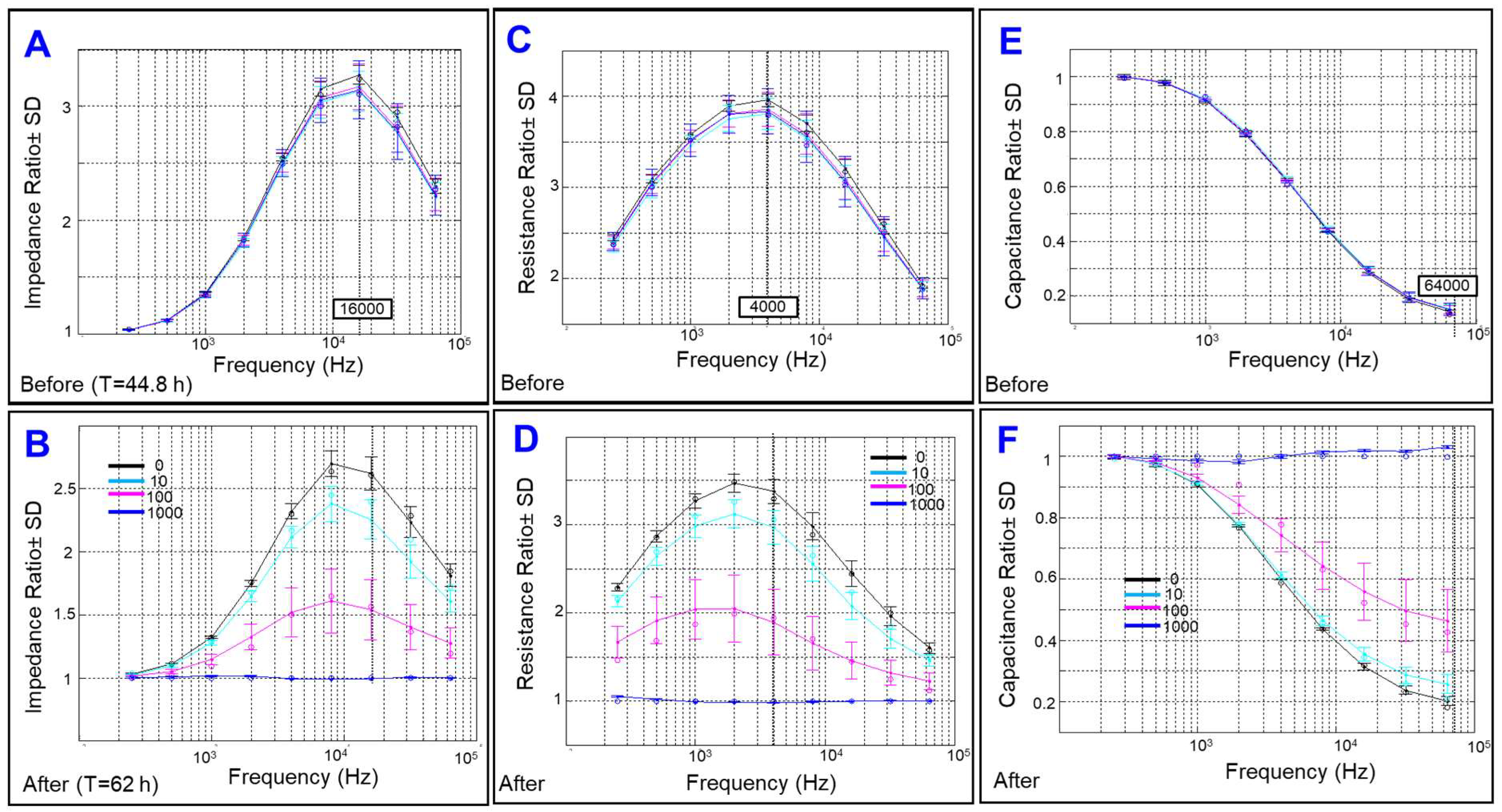
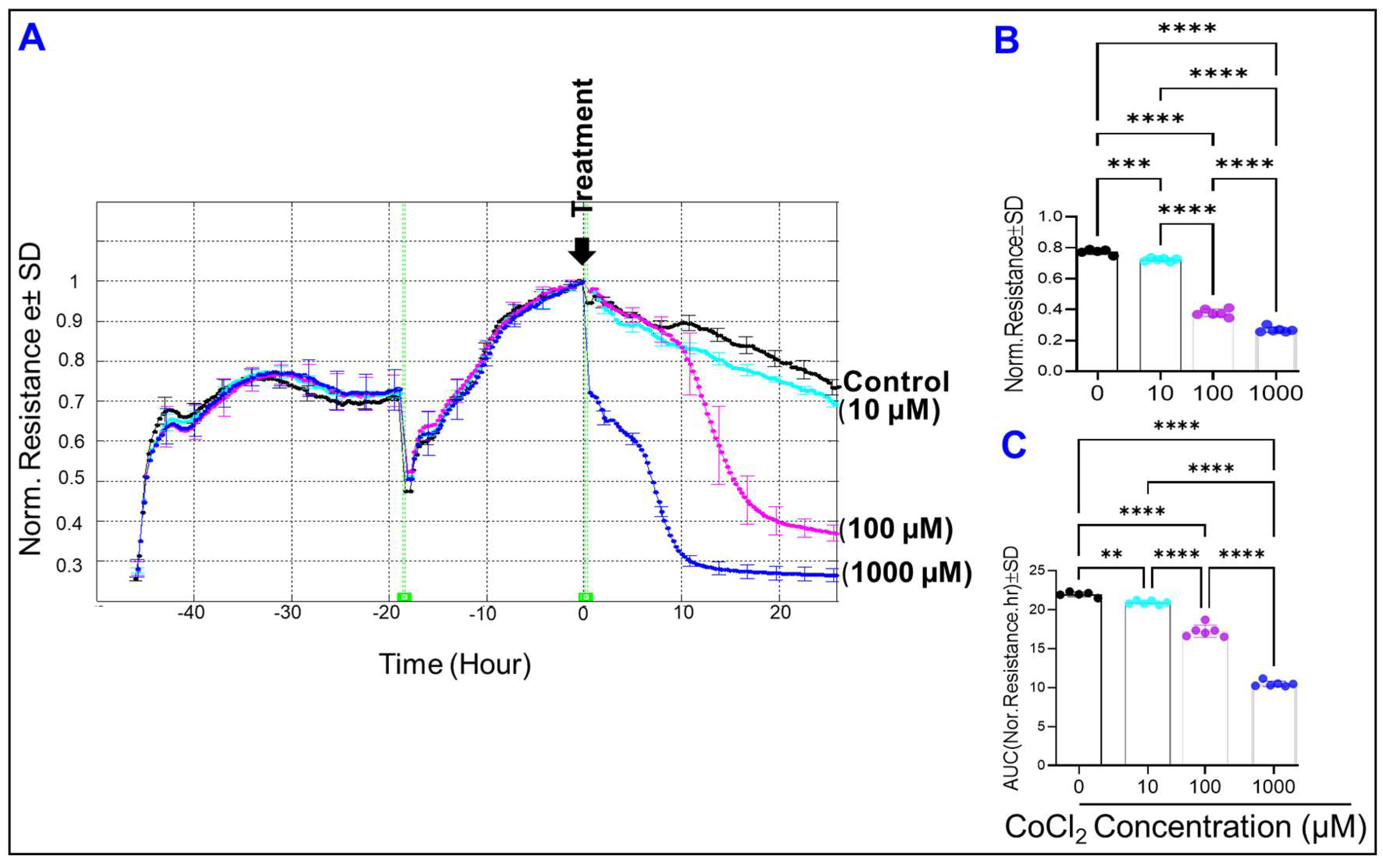
2.1. Effect of Cytopathic Hypoxia on HREC Electrical Impedance
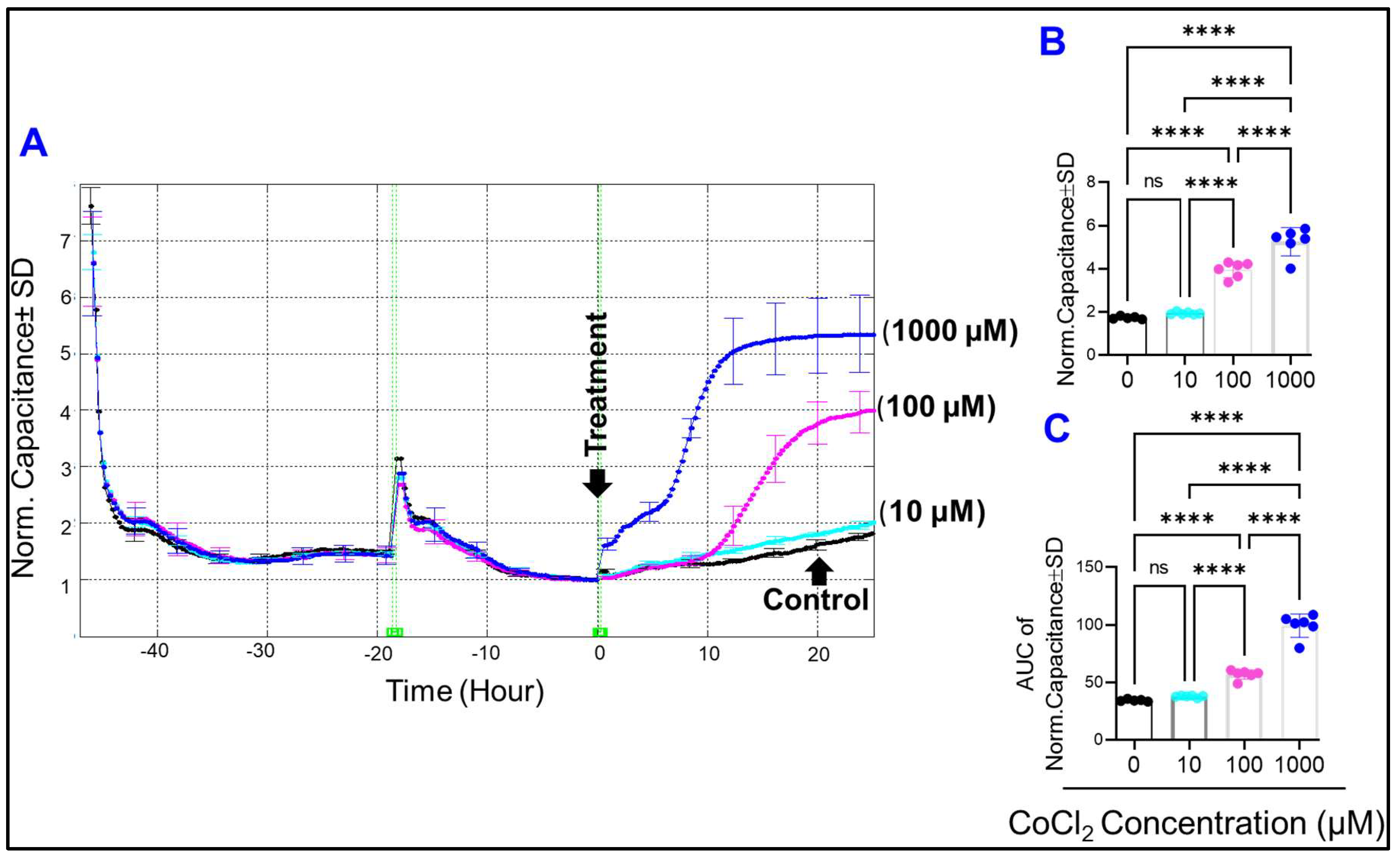
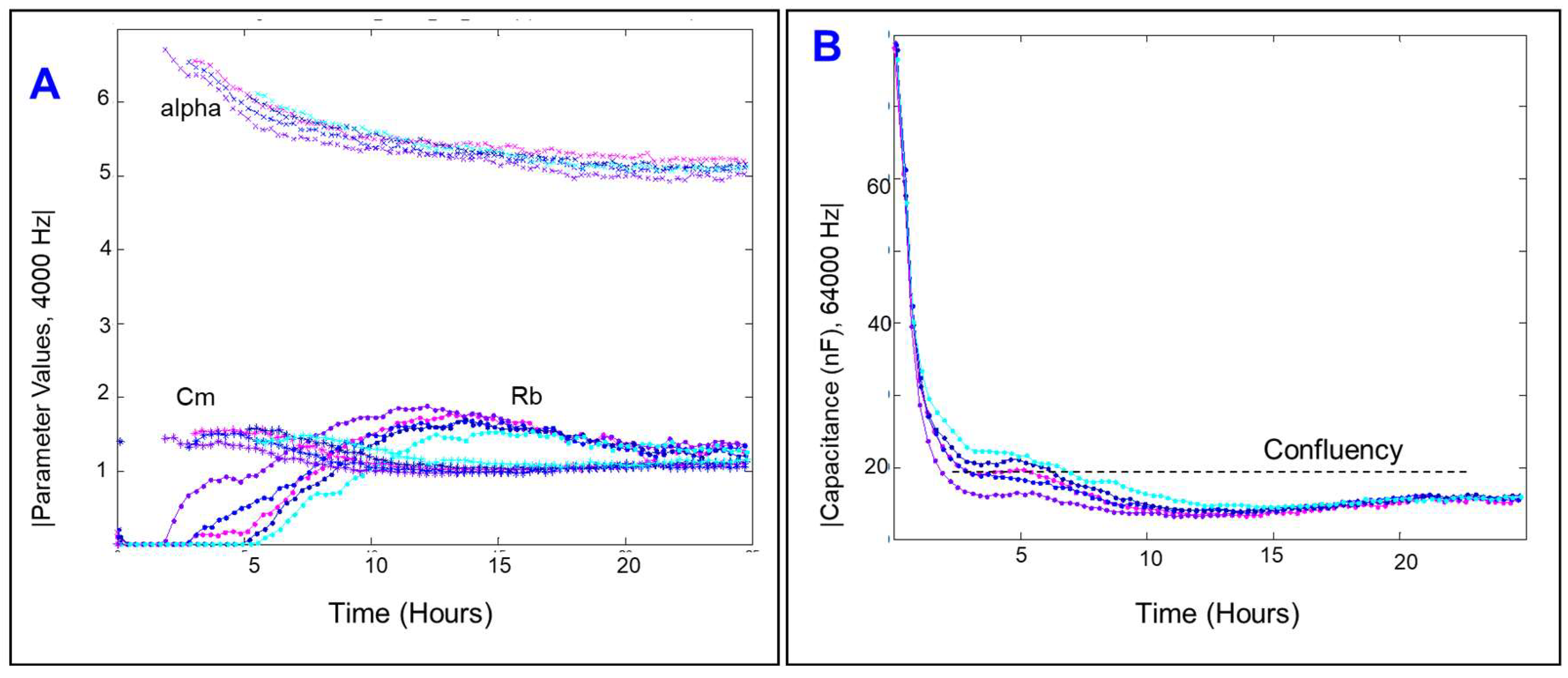
2.2. Effect of Cytopathic Hypoxia on the Capacitance of HRECs
2.3. Effect of Cytopathic Hypoxia on the Total Resistance of HRECs
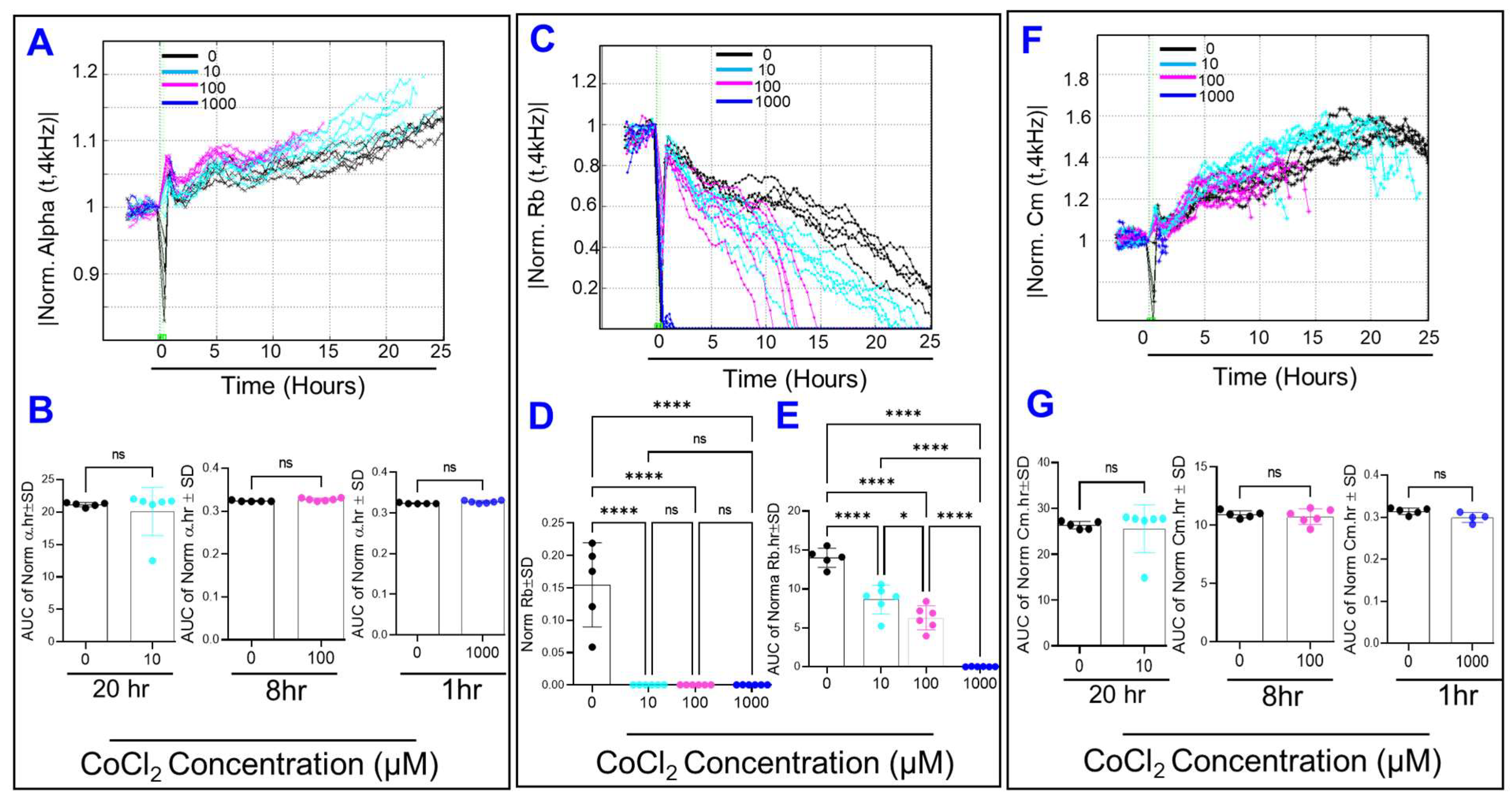
2.4. Effects of Cytopathic Hypoxia on Components of Transendothelial Resistance of HRECs
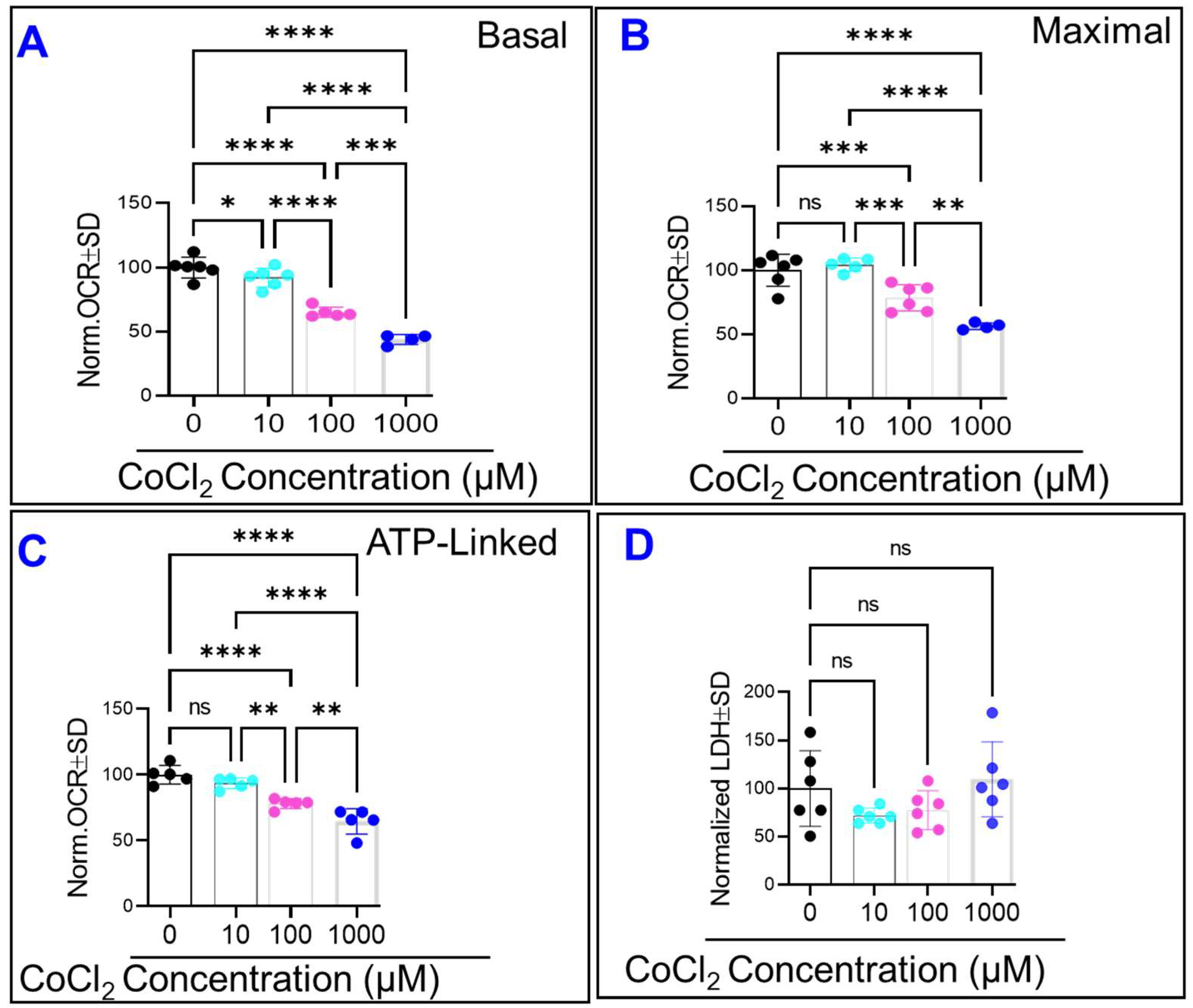
2.5. CoCl2 Compromises HRECs’ Mitochondrial Bioenergetics without Causing Cytotoxicity
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. ECIS Experiment and Modeling
5.2. Assessment of HREC Viability
5.3. Mitochondrial Bioenergetic Profiles
5.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-retinal barrier. Eur. J. Ophthalmol. 2011, 21 (Suppl. 6), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.S.; Appukuttan, B.; Wilmarth, P.A.; Pan, Y.; Stempel, A.J.; Chipps, T.J.; Benedetti, E.E.; Zamora, D.O.; Choi, D.; David, L.L.; et al. Role of the retinal vascular endothelial cell in ocular disease. Prog. Retin Eye Res. 2013, 32, 102–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshet, E. More weapons in the arsenal against ischemic retinopathy. J. Clin. Investig. 2001, 107, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.C.; Dabouz, R.; Noueihed, B.; Omri, S.; Tahiri, H.; Chemtob, S. Ischemic Retinopathies: Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2017, 2017, 3940241. [Google Scholar] [CrossRef] [Green Version]
- Jampol, L.M.; Glassman, A.R.; Sun, J. Evaluation and Care of Patients with Diabetic Retinopathy. N. Engl. J. Med. 2020, 382, 1629–1637. [Google Scholar] [CrossRef]
- Al-Latayfeh, M.; Silva, P.S.; Sun, J.K.; Aiello, L.P. Antiangiogenic therapy for ischemic retinopathies. Cold Spring Harb. Perspect. Med. 2012, 2, a006411. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.D.; Lindsley, K.; Vedula, S.S.; Krzystolik, M.G.; Hawkins, B.S. Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2019, 3, CD005139. [Google Scholar] [CrossRef]
- Virgili, G.; Parravano, M.; Evans, J.R.; Gordon, I.; Lucenteforte, E. Anti-vascular endothelial growth factor for diabetic macular oedema: A network meta-analysis. Cochrane Database Syst. Rev. 2018, 10, CD007419. [Google Scholar] [CrossRef] [Green Version]
- D’Amore, P.A. Vascular endothelial cell growth factor-a: Not just for endothelial cells anymore. Am. J. Pathol. 2007, 171, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Nair, P.; Aiello, L.P.; Gardner, T.W.; Jampol, L.M.; Ferris, F.L., III. Report From the NEI/FDA Diabetic Retinopathy Clinical Trial Design and Endpoints Workshop. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5127–5142. [Google Scholar] [CrossRef]
- Wright, C.; Mazzucco, A.E.; Becker, S.M.; Sieving, P.A.; Tumminia, S.J. NEI-Supported Age-Related Macular Degeneration Research: Past, Present, and Future. Transl. Vis. Sci. Technol. 2020, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Shosha, E.; Fouda, A.Y.; Lemtalsi, T.; Haigh, S.; Fulton, D.; Ibrahim, A.; Al-Shabrawey, M.; Caldwell, R.W.; Caldwell, R.B. Endothelial arginase 2 mediates retinal ischemia/reperfusion injury by inducing mitochondrial dysfunction. Mol. Metab. 2021, 53, 101273. [Google Scholar] [CrossRef] [PubMed]
- Heyck, M.; Bonsack, B.; Zhang, H.; Sadanandan, N.; Cozene, B.; Kingsbury, C.; Lee, J.Y.; Borlongan, C.V. The brain and eye: Treating cerebral and retinal ischemia through mitochondrial transfer. Exp. Biol. Med. 2019, 244, 1485–1492. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Lai, T.T.; Li, C.J. The Oxidative Stress and Mitochondrial Dysfunction during the Pathogenesis of Diabetic Retinopathy. Oxid. Med. Cell Longev. 2018, 2018, 3420187. [Google Scholar] [CrossRef] [PubMed]
- Caltana, L.; Merelli, A.; Lazarowski, A.; Brusco, A. Neuronal and glial alterations due to focal cortical hypoxia induced by direct cobalt chloride (CoCl2) brain injection. Neurotox. Res. 2009, 15, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Niwa, M.; Aoki, H.; Kumada, M.; Kunisada, T.; Oyama, T.; Yamamoto, T.; Kozawa, O.; Mori, H. A new model of retinal photoreceptor cell degeneration induced by a chemical hypoxia-mimicking agent, cobalt chloride. Brain Res. 2006, 1109, 192–200. [Google Scholar] [CrossRef]
- Munoz-Sanchez, J.; Chanez-Cardenas, M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 71–103. [Google Scholar] [CrossRef]
- Diaz-Coranguez, M.; Ramos, C.; Antonetti, D.A. The inner blood-retinal barrier: Cellular basis and development. Vis. Res. 2017, 139, 123–137. [Google Scholar] [CrossRef]
- Guerra, M.H.; Yumnamcha, T.; Ebrahim, A.S.; Berger, E.A.; Singh, L.P.; Ibrahim, A.S. Real-Time Monitoring the Effect of Cytopathic Hypoxia on Retinal Pigment Epithelial Barrier Functionality Using Electric Cell-Substrate Impedance Sensing (ECIS) Biosensor Technology. Int. J. Mol. Sci. 2021, 22, 456822. [Google Scholar] [CrossRef]
- Giaever, I.; Keese, C.R. A morphological biosensor for mammalian cells. Nature 1993, 366, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Robilliard, L.D.; Kho, D.T.; Johnson, R.H.; Anchan, A.; O’Carroll, S.J.; Graham, E.S. The Importance of Multifrequency Impedance Sensing of Endothelial Barrier Formation Using ECIS Technology for the Generation of a Strong and Durable Paracellular Barrier. Biosensors 2018, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslin, J.W.; Kurtz, K.M. Lymphatic endothelial cells adapt their barrier function in response to changes in shear stress. Lymphat. Res. Biol. 2009, 7, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Orsenigo, F.; Molendini, C.; Baluk, P.; McDonald, D.M. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009, 335, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, S.; Al-Ahmad, A.J.; Gassmann, M.; Ogunshola, O.O. Hypoxia selectively disrupts brain microvascular endothelial tight junction complexes through a hypoxia-inducible factor-1 (HIF-1) dependent mechanism. J. Cell Physiol. 2014, 229, 1096–1105. [Google Scholar] [CrossRef]
- Balaiya, S.; Murthy, R.K.; Chalam, K.V. Resveratrol inhibits proliferation of hypoxic choroidal vascular endothelial cells. Mol. Vis. 2013, 19, 2385–2392. [Google Scholar]
- Pedram, A.; Razandi, M.; Levin, E.R. Deciphering vascular endothelial cell growth factor/vascular permeability factor signaling to vascular permeability. Inhibition by atrial natriuretic peptide. J. Biol. Chem. 2002, 277, 44385–44398. [Google Scholar] [CrossRef] [Green Version]
- Al Ahmad, A.; Gassmann, M.; Ogunshola, O.O. Maintaining blood-brain barrier integrity: Pericytes perform better than astrocytes during prolonged oxygen deprivation. J. Cell Physiol. 2009, 218, 612–622. [Google Scholar] [CrossRef]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, G.; Khan, S.A.; Luo, W.; Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell Physiol. 2011, 226, 2925–2933. [Google Scholar] [CrossRef] [Green Version]
- Haidari, M.; Zhang, W.; Wakame, K. Disruption of endothelial adherens junction by invasive breast cancer cells is mediated by reactive oxygen species and is attenuated by AHCC. Life Sci. 2013, 93, 994–1003. [Google Scholar] [CrossRef]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R., Jr.; Ballinger, S.; Dell’Italia, L.; Jianhua, Z.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Izumi, Y.; Nagashima, Y.; Tamai-Nagai, Y.; Kurihara, H.; Sakai, T.; Suzuki, Y.; Yamanaka, T.; Suzuki, A.; Mizuno, K.; et al. Involvement of ASIP/PAR-3 in the promotion of epithelial tight junction formation. J. Cell Sci. 2002, 115, 2485–2495. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Hallett, M.A.; Atkinson, S.J.; Marrs, J.A. aPKC-PAR complex dysfunction and tight junction disassembly in renal epithelial cells during ATP depletion. Am. J. Physiol. Cell Physiol. 2007, 292, C1094–C1102. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Hussein, K.; Wang, F.; Wan, M.; Saad, N.; Essa, M.; Kim, I.; Shakoor, A.; Owen, L.A.; DeAngelis, M.M.; et al. Bone Morphogenetic Protein (BMP)4 But Not BMP2 Disrupts the Barrier Integrity of Retinal Pigment Epithelia and Induces Their Migration: A Potential Role in Neovascular Age-Related Macular Degeneration. J. Clin. Med. 2020, 9, 2293. [Google Scholar] [CrossRef]
- Guerra, M.H.; Yumnamcha, T.; Singh, L.P.; Ibrahim, A.S. Relative Contribution of Different Mitochondrial Oxidative Phosphorylation Components to the Retinal Pigment Epithelium Barrier Function: Implications for RPE-Related Retinal Diseases. Int. J. Mol. Sci 2021, 22, 8130. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-tanani, S.; Yumnamcha, T.; Singh, L.P.; Ibrahim, A.S. Differential Effects of Cytopathic Hypoxia on Human Retinal Endothelial Cellular Behavior: Implication for Ischemic Retinopathies. Int. J. Mol. Sci. 2022, 23, 4274. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084274
El-tanani S, Yumnamcha T, Singh LP, Ibrahim AS. Differential Effects of Cytopathic Hypoxia on Human Retinal Endothelial Cellular Behavior: Implication for Ischemic Retinopathies. International Journal of Molecular Sciences. 2022; 23(8):4274. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084274
Chicago/Turabian StyleEl-tanani, Shaimaa, Thangal Yumnamcha, Lalit Pukhrambam Singh, and Ahmed S. Ibrahim. 2022. "Differential Effects of Cytopathic Hypoxia on Human Retinal Endothelial Cellular Behavior: Implication for Ischemic Retinopathies" International Journal of Molecular Sciences 23, no. 8: 4274. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084274