
PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients


Abstract
:1. Introduction
2. PPAR Agonists in the Treatment of NAFLD with Concomitant T2DM
2.1. Molecular Basics of PPAR-Dependent Regulation
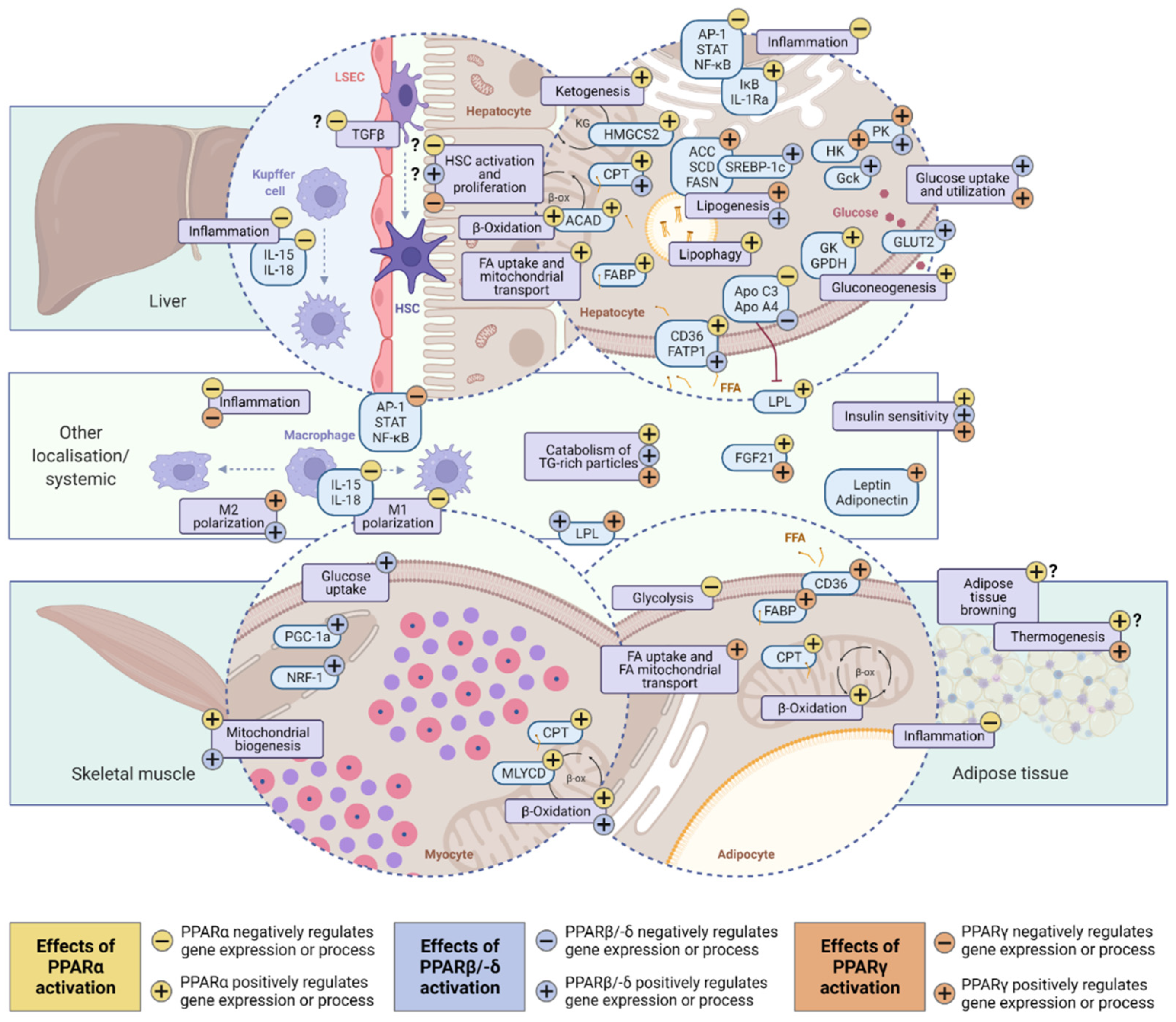
2.2. PPARα (NR1C1)
2.3. PPARδ (PPARβ; NR1C2)
2.4. PPARγ (NR1C3)
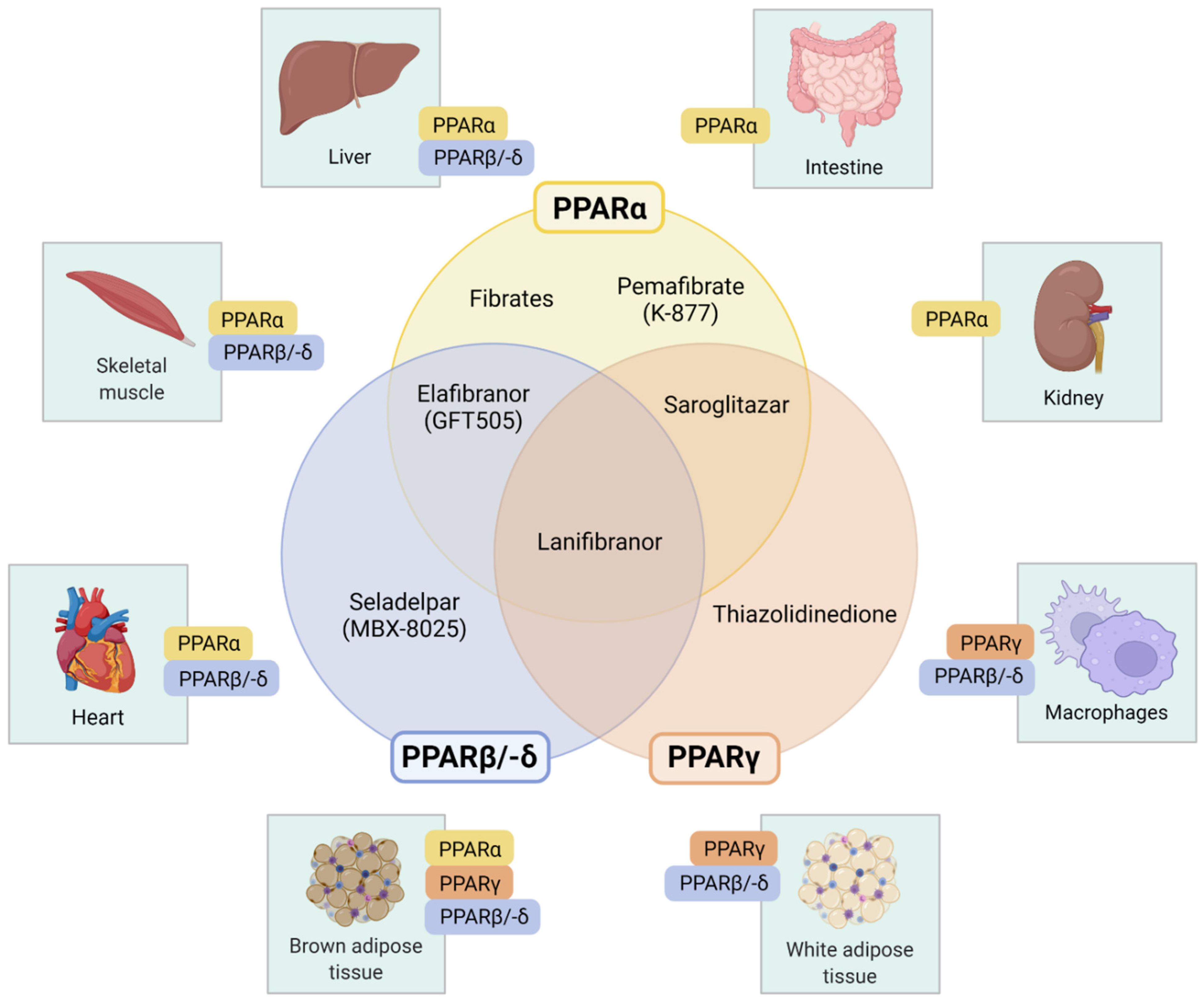
3. Pharmacologic PPAR-Targeted Therapies
3.1. Selective PPARα Modulator: Pemafibrate (K-877)
3.2. PPARα Agonists: Fibrates
3.3. Selective PPARδ/β Agonist: Seladelpar (MBX-8025)
3.4. PPARγ Agonists: Thiazolidinediones
3.4.1. Pioglitazone
3.4.2. Rosiglitazone
3.4.3. Lobeglitazone
3.5. Dual PPARα and -γ Agonist: Saroglitazar
3.6. Dual PPARα and -δ Agonist: Elafibranor (GFT505)
3.7. Pan-PPAR-Agonist: Lanifibranor (IVA337)
4. Comorbidities of the Metabolic Syndrome in the PPAR-Targeted Treatment of Diabetic NAFLD Patients
4.1. Overweight and Obesity
4.2. Dyslipidemia
4.3. Cardiovascular Comorbidities
5. Outlook and Further Areas of Research
6. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019, 1, 312–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef]
- Kanwal, F.; Shubrook, J.H.; Younossi, Z.; Natarajan, Y.; Bugianesi, E.; Rinella, M.E. Preparing for the NASH Epidemic: A Call to Action. Gastroenterology 2021, 161, 1030–1042.e8. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Tran, T.; Everhart, J.E. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef]
- Noureddin, N.; Noureddin, M.; Singh, A.; Alkhouri, N. Progression of Nonalcoholic Fatty Liver Disease-Associated Fibrosis in a Large Cohort of Patients with Type 2 Diabetes. Dig. Dis. Sci. 2022, 67, 1379–1388. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Bonora, E.; Targher, G. Nonalcoholic Fatty Liver Disease and Risk of Incident Type 2 Diabetes: A Meta-analysis. Diabetes Care 2018, 41, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Bril, F. Nonalcoholic fatty liver disease in type 2 diabetes: Awareness is the first step toward change. Hepatobiliary Surg. Nutr. 2020, 9, 493–496. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Lalloyer, F.; Staels, B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arter. Thromb. Vasc. Biol. 2010, 30, 894–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroy-Ramirez, H.C.; Galicia-Moreno, M.; Sandoval-Rodriguez, A.; Meza-Rios, A.; Santos, A.; Armendariz-Borunda, J. PPARs as Metabolic Sensors and Therapeutic Targets in Liver Diseases. Int. J. Mol. Sci. 2021, 22, 8298. [Google Scholar] [CrossRef] [PubMed]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.-F. Nonalcoholic steatohepatitis: The role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2007, 1771, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α. Future Cardiol. 2017, 13, 259–278. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tailleux, A.; Wouters, K.; Staels, B. Roles of PPARs in NAFLD: Potential therapeutic targets. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2011, 1821, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Régnier, M.; Polizzi, A.; Lippi, Y.; Fouché, E.; Michel, G.; Lukowicz, C. Insights into the role of hepatocyte PPARα activity in response to fasting. Mol. Cell. Endocrinol. 2018, 471, 75–88. [Google Scholar] [CrossRef]
- Ribet, C.; Montastier, E.; Valle, C.; Bezaire, V.; Mazzucotelli, A.; Mairal, A. Peroxisome proliferator-activated receptor-alpha control of lipid and glucose metabolism in human white adipocytes. Endocrinology 2010, 151, 123–133. [Google Scholar] [CrossRef]
- Muoio, D.M.; Way, J.M.; Tanner, C.J.; Winegar, D.A.; Kliewer, S.A.; Houmard, J.A. Peroxisome proliferator-activated receptor-alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002, 51, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, M.; Baugé, E.; Bourguet, W.; De Bosscher, K.; Lalloyer, F.; Tailleux, A. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology 2014, 60, 1593–1606. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.; Hall, P.; Robertson, G.; Leclercq, I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 2004, 39, 1286–1296. [Google Scholar] [CrossRef]
- Chen, L.; Li, L.; Chen, J.; Li, L.; Zheng, Z.; Ren, J. Oleoylethanolamide, an endogenous PPAR-α ligand, attenuates liver fibrosis targeting hepatic stellate cells. Oncotarget 2015, 6, 42530–42540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smati, S.; Polizzi, A.; Fougerat, A.; Ellero-Simatos, S.; Blum, Y.; Lippi, Y. Integrative study of diet-induced mouse models of NAFLD identifies PPARα as a sexually dimorphic drug target. Gut 2021, 71, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, N.; Pradervand, S.; Wahli, W. Sumoylated PPARalpha mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J. Clin. Investig. 2009, 119, 3138–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Downes, M.; Yu, R.T.; Bookout, A.L.; He, W.; Straume, M. Nuclear receptor expression links the circadian clock to metabolism. Cell 2006, 126, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Montagner, A.; Tan, N.S.; Wahli, W. Insights into the Role of PPARβ/δ in NAFLD. Int. J. Mol. Sci. 2018, 19, 1893. [Google Scholar] [CrossRef] [Green Version]
- Zarei, M.; Aguilar-Recarte, D.; Palomer, X.; Vázquez-Carrera, M. Revealing the role of peroxisome proliferator-activated receptor β/δ in nonalcoholic fatty liver disease. Metabolism 2021, 114, 154342. [Google Scholar] [CrossRef]
- Takahashi, S.; Tanaka, T.; Kodama, T.; Sakai, J. Peroxisome proliferator-activated receptor delta (PPARdelta), a novel target site for drug discovery in metabolic syndrome. Pharmacol. Res. 2006, 53, 501–507. [Google Scholar] [CrossRef]
- Seedorf, U.; Aberle, J. Emerging roles of PPARdelta in metabolism. Biochim. Biophys. Acta 2007, 1771, 1125–1131. [Google Scholar] [CrossRef]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Peña, L.; Botteri, G.; Zarei, M. PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-β/δ and PPAR-γ. Future Cardiol. 2017, 13, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor β/δ in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.-M.; Tardivel, A. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef]
- Koh, J.-H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.-H. PPARβ Is Essential for Maintaining Normal Levels of PGC-1α and Mitochondria and for the Increase in Muscle Mitochondria Induced by Exercise. Cell Metab. 2017, 25, 1176–1185.e5. [Google Scholar] [CrossRef]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.-C.; Kang, K.; Reilly, S.M. Role of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Brown, J.D.; Stanya, K.J.; Homan, E.; Leidl, M.; Inouye, K. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, L.M.; Boekschoten, M.V.; Desvergne, B.; Müller, M.; Kersten, S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol. Genom. 2010, 41, 42–52. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef] [Green Version]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.e6. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the Roles of PPARγ in Adipocytes via Dynamic Change of Transcription Complex. Front. Endocrinol. 2018, 9, 473. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Mullican, S.E.; DiSpirito, J.R.; Peed, L.C.; Lazar, M.A. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. Proc. Natl. Acad. Sci. USA 2013, 110, 18656–18661. [Google Scholar] [CrossRef] [Green Version]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Takahashi, M.; Funahashi, T.; Kihara, S.; Nishizawa, H.; Kishida, K. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 2001, 50, 2094–2099. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Hazra, S.; Xiong, S.; Wang, J.; Rippe, R.A.; Krishna, V.; Chatterjee, K. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J. Biol. Chem. 2004, 279, 11392–11401. [Google Scholar] [CrossRef] [Green Version]
- Galli, A.; Crabb, D.W.; Ceni, E.; Salzano, R.; Mello, T.; Svegliati–Baroni, G. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology 2002, 122, 1924–1940. [Google Scholar] [CrossRef]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharm. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef]
- Yokote, K.; Yamashita, S.; Arai, H.; Araki, E.; Matsushita, M.; Nojima, T. Effects of pemafibrate on glucose metabolism markers and liver function tests in patients with hypertriglyceridemia: A pooled analysis of six phase 2 and phase 3 randomized double-blind placebo-controlled clinical trials. Cardiovasc. Diabetol. 2021, 20, 96. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Byrne, C.D.; Scorletti, E.; Mantzoros, C.; Targher, G. Efficacy and safety of anti-hyperglycaemic drugs in patients with non-alcoholic fatty liver disease with or without diabetes: An updated systematic review of randomized controlled trials. Diabetes Metab. 2020, 46, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J. Role of Vitamin E for Nonalcoholic Steatohepatitis in Patients with Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C. Long-Term Pioglitazone Treatment for Patients with Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.-C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients with Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 2018, 16, 558–566.e2. [Google Scholar] [CrossRef]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K. Saroglitazar, a PPAR-α/γ Agonist, for Treatment of Nonalcoholic Fatty Liver Disease: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and-δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [Green Version]
- Raza-Iqbal, S.; Tanaka, T.; Anai, M.; Inagaki, T.; Matsumura, Y.; Ikeda, K. Transcriptome Analysis of K-877 (a Novel Selective PPARα Modulator (SPPARMα))-Regulated Genes in Primary Human Hepatocytes and the Mouse Liver. J. Atheroscler. Thromb. 2015, 22, 754–772. [Google Scholar] [CrossRef] [Green Version]
- Fruchart, J.-C.; Santos, R.D.; Aguilar-Salinas, C.; Aikawa, M.; Al Rasadi, K.; Amarenco, P. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMα) paradigm: Conceptual framework and therapeutic potential: A consensus statement from the International Atherosclerosis Society (IAS) and the Residual Risk Reduction Initiative (R3i) Foundation. Cardiovasc. Diabetol. 2019, 18, 71. [Google Scholar]
- Ida, S.; Kaneko, R.; Murata, K. Efficacy and safety of pemafibrate administration in patients with dyslipidemia: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Asahiyama, M.; Tanaka, T.; Yamamoto, S.; Murakami, K.; Kamiya, W. Pemafibrate, a selective PPARα modulator, prevents non-alcoholic steatohepatitis development without reducing the hepatic triglyceride content. Sci. Rep. 2020, 10, 7818. [Google Scholar] [CrossRef]
- Ishibashi, S.; Yamashita, S.; Arai, H.; Araki, E.; Yokote, K.; Suganami, H. Effects of K-877, a novel selective PPARα modulator (SPPARMα), in dyslipidaemic patients: A randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 2016, 249, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T. Effects of Pemafibrate, a Novel Selective PPARα Modulator, on Lipid and Glucose Metabolism in Patients with Type 2 Diabetes and Hypertriglyceridemia: A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial. Diabetes Care 2018, 41, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor α modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Seko, Y.; Yamaguchi, K.; Umemura, A.; Yano, K.; Takahashi, A.; Okishio, S. Effect of pemafibrate on fatty acid levels and liver enzymes in non-alcoholic fatty liver disease patients with dyslipidemia: A single-arm, pilot study. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2020, 50, 1328–1336. [Google Scholar] [CrossRef]
- Hatanaka, T.; Kosone, T.; Saito, N.; Takakusagi, S.; Tojima, H.; Naganuma, A. Effect of 48-week pemafibrate on non-alcoholic fatty liver disease with hypertriglyceridemia, as evaluated by the FibroScan-aspartate aminotransferase score. JGH Open 2021, 5, 1183–1189. [Google Scholar] [CrossRef]
- Hatanaka, T.; Kakizaki, S.; Saito, N.; Nakano, Y.; Nakano, S.; Hazama, Y. Impact of Pemafibrate in Patients with Hypertriglyceridemia and Metabolic Dysfunction-associated Fatty Liver Disease Pathologically Diagnosed with Non-alcoholic Steatohepatitis: A Retrospective, Single-arm Study. Intern. Med. Tokyo Jpn. 2021, 2021, 6574-20. [Google Scholar] [CrossRef]
- Shinozaki, S.; Tahara, T.; Lefor, A.K.; Ogura, M. Pemafibrate decreases markers of hepatic inflammation in patients with non-alcoholic fatty liver disease. Clin. Exp. Hepatol. 2020, 6, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Tahara, T.; Lefor, A.K.; Ogura, M. Pemafibrate improves hepatic inflammation, function and fibrosis in patients with non-alcoholic fatty liver disease: A one-year observational study. Clin. Exp. Hepatol. 2021, 7, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: A pilot study. Hepatology 1996, 23, 1464–1467. [Google Scholar] [CrossRef]
- Honda, A.; Tanaka, A.; Kaneko, T.; Komori, A.; Abe, M.; Inao, M. Bezafibrate Improves GLOBE and UK-PBC Scores and Long-Term Outcomes in Patients With Primary Biliary Cholangitis. Hepatology 2019, 70, 2035–2046. [Google Scholar] [CrossRef]
- Feng, X.; Gao, X.; Jia, Y.; Zhang, H.; Xu, Y.; Wang, G. PPAR-α Agonist Fenofibrate Decreased RANTES Levels in Type 2 Diabetes Patients with Hypertriglyceridemia. Med. Sci. Monit. 2016, 22, 743–751. [Google Scholar] [CrossRef]
- Li, B.-H.; He, F.-P.; Yang, X.; Chen, Y.-W.; Fan, J.-G. Steatosis induced CCL5 contributes to early-stage liver fibrosis in nonalcoholic fatty liver disease progress. Transl. Res. 2017, 180, 103–117.e4. [Google Scholar] [CrossRef]
- Nikam, A.; Patankar, J.V.; Somlapura, M.; Lahiri, P.; Sachdev, V.; Kratky, D. The PPARα Agonist Fenofibrate Prevents Formation of Protein Aggregates (Mallory-Denk bodies) in a Murine Model of Steatohepatitis-like Hepatotoxicity. Sci. Rep. 2018, 8, 12964. [Google Scholar] [CrossRef]
- Rodríguez-Vilarrupla, A.; Laviña, B.; García-Calderó, H.; Russo, L.; Rosado, E.; Roglans, N. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J. Hepatol. 2012, 56, 1033–10369. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Berria, R.; Cornell, J.; Cusi, K. Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 829–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, E.; Mohammed, B.S.; Korenblat, K.M.; Magkos, F.; McCrea, J.; Patterson, B.W. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2010, 95, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Oscarsson, J.; Önnerhag, K.; Risérus, U.; Sundén, M.; Johansson, L.; Jansson, P.-A. Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: A double-blind, randomized, placebo-controlled study. J. Clin. Lipidol. 2018, 12, 1390–1403.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karhapää, P.; Uusitupa, M.; Voutilainen, E.; Laakso, M. Effects of bezafibrate on insulin sensitivity and glucose tolerance in subjects with combined hyperlipidemia. Clin. Pharmacol. Ther. 1992, 52, 620–626. [Google Scholar] [CrossRef]
- LiverTox. Fibrates. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK547893/ (accessed on 10 October 2021).
- Jakob, T.; Nordmann, A.J.; Schandelmaier, S.; Ferreira-González, I.; Briel, M. Fibrates for primary prevention of cardiovascular disease events. Cochrane Database Syst. Rev. 2016, 11, CD009753. [Google Scholar] [CrossRef]
- Bays, H.E.; Schwartz, S.; Littlejohn, T.; Kerzner, B.; Krauss, R.M.; Karpf, D.B. MBX-8025, a novel peroxisome proliferator receptor-delta agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Chiquette, E.; Ramirez, G.; Defronzo, R. A meta-analysis comparing the effect of thiazolidinediones on cardiovascular risk factors. Arch. Intern. Med. 2004, 164, 2097–2104. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, E.; Csako, G.; Pucino, F.; Wesley, R.; Loomba, R. Meta-analysis: Pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2012, 35, 66–75. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Liu, X.; Wang, L.; Yang, Z. Thiazolidinediones for nonalcoholic steatohepatitis: A meta-analysis of randomized clinical trials. Medicine 2016, 95, e4947. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.; Kimura, H.; Moriyama, S.; Odaka, H.; Momose, Y.; Sugiyama, Y. Activation of human peroxisome proliferator-activated receptor (PPAR) subtypes by pioglitazone. Biochem. Biophys. Res. Commun. 2000, 278, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Duseja, A.; Singh, S.P.; Saraswat, V.A.; Acharya, S.K.; Chawla, Y.K.; Chowdhury, S. Non-alcoholic Fatty Liver Disease and Metabolic Syndrome-Position Paper of the Indian National Association for the Study of the Liver, Endocrine Society of India, Indian College of Cardiology and Indian Society of Gastroenterology. J. Clin. Exp. Hepatol. 2015, 5, 51–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.H.; Lee, H.W.; Yoo, J.-J.; Cho, Y.; Kim, S.U.; Lee, T.H. KASL clinical practice guidelines: Management of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2021, 27, 363–401. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Dirchwolf, M.; Álvares-da-Silva, M.R.; Barrera, F.; Benítez, C.; Castellanos-Fernandez, M. Latin American Association for the study of the liver (ALEH) practice guidance for the diagnosis and treatment of non-alcoholic fatty liver disease. Ann. Hepatol. 2020, 19, 674–690. [Google Scholar] [CrossRef]
- EASL–EASD–EASO. EASL–EASD–EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Panunzi, S.; Maltese, S.; Verrastro, O.; Labbate, L.; De Gaetano, A.; Pompili, M. Pioglitazone and bariatric surgery are the most effective treatments for non-alcoholic steatohepatitis: A hierarchical network meta-analysis. Diabetes Obes. Metab. 2021, 23, 980–990. [Google Scholar] [CrossRef]
- Hsiao, P.-J.; Chiou, H.-Y.C.; Jiang, H.-J.; Lee, M.-Y.; Hsieh, T.-J.; Kuo, K.-K. Pioglitazone Enhances Cytosolic Lipolysis, β-oxidation and Autophagy to Ameliorate Hepatic Steatosis. Sci. Rep. 2017, 7, 9030. [Google Scholar] [CrossRef] [Green Version]
- de Mendonça, M.; Dos Santos, B.D.A.C.; de Sousa, É.; Rodrigues, A.C. Adiponectin is required for pioglitazone-induced improvements in hepatic steatosis in mice fed a high-fat diet. Mol. Cell. Endocrinol. 2019, 493, 110480. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Miyazaki, Y.; Mahankali, A.; Berria, R.; Pettiti, M.; Buzzigoli, E. The effect of pioglitazone on the liver: Role of adiponectin. Diabetes Care 2006, 29, 2275–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, I.A.; Sempoux, C.; Stärkel, P.; Horsmans, Y. Limited therapeutic efficacy of pioglitazone on progression of hepatic fibrosis in rats. Gut 2006, 55, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi-Suzuki, M.; Cusi, K.; Bril, F.; Gong, Y.; Langaee, T.; Frye, R.F. A Genetic Score Associates with Pioglitazone Response in Patients With Non-alcoholic Steatohepatitis. Front. Pharmacol. 2018, 9, 752. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Sanyal, A.J.; Neuschwander-Tetri, B.A. Improvements in Histologic Features and Diagnosis Associated with Improvement in Fibrosis in Nonalcoholic Steatohepatitis: Results from the Nonalcoholic Steatohepatitis Clinical Research Network Treatment Trials. Hepatology 2019, 70, 522–531. [Google Scholar] [PubMed]
- Chalasani, N.P.; Sanyal, A.J.; Kowdley, K.V.; Robuck, P.R.; Hoofnagle, J.; Kleiner, D.E. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp. Clin. Trials 2009, 30, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J. A Placebo-Controlled Trial of Pioglitazone in Subjects with Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Fu, J. Pioglitazone for NAFLD Patients with Prediabetes or Type 2 Diabetes Mellitus: A Meta-Analysis. Front. Endocrinol. 2021, 12, 615409. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Inzucchi, S.; Abdul-Ghani, M.; Nissen, S.E. Pioglitazone: The forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diab. Vasc. Dis. Res. 2019, 16, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Defronzo, R.A.; Tripathy, D.; Schwenke, D.C.; Banerji, M.; Bray, G.A.; Buchanan, T.A. Prevention of diabetes with pioglitazone in ACT NOW: Physiologic correlates. Diabetes 2013, 62, 3920–3926. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D.; Schwenke, D.C.; Banerji, M.; Bray, G.A.; Buchanan, T.A. Pioglitazone for diabetes prevention in impaired glucose tolerance. N. Engl. J. Med. 2011, 364, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Viscoli, C.M.; Young, L.H.; Furie, K.L.; Gorman, M.; Lovejoy, A.M. Pioglitazone Prevents Diabetes in Patients with Insulin Resistance and Cerebrovascular Disease. Diabetes Care 2016, 39, 1684–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Shi, W.; Fu, S.; Wang, T.; Zhai, S.; Song, Y. Pioglitazone and bladder cancer risk: A systematic review and meta-analysis. Cancer Med. 2018, 7, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Ripamonti, E.; Azoulay, L.; Abrahamowicz, M.; Platt, R.; Suissa, S. A systematic review of observational studies of the association between pioglitazone use and bladder cancer. Diabet. Med. 2018, 36, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Filipova, E.; Uzunova, K.; Kalinov, K.; Vekov, T. Pioglitazone and the Risk of Bladder Cancer: A Meta-Analysis. Diabetes Ther. 2017, 8, 705–726. [Google Scholar] [CrossRef]
- Portillo-Sanchez, P.; Bril, F.; Lomonaco, R.; Barb, D.; Orsak, B.; Bruder, J.M. Effect of pioglitazone on bone mineral density in patients with nonalcoholic steatohepatitis: A 36-month clinical trial. J. Diabetes 2019, 11, 223–231. [Google Scholar] [CrossRef]
- Tuccori, M.; Filion, K.B.; Yin, H.; Yu, O.H.; Platt, R.W.; Azoulay, L. Pioglitazone use and risk of bladder cancer: Population based cohort study. BMJ 2016, 352, i1541. [Google Scholar] [CrossRef] [Green Version]
- Neuschwander-Tetri, B.A.; Brunt, E.M.; Wehmeier, K.R.; Oliver, D.; Bacon, B.R. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology 2003, 38, 1008–1017. [Google Scholar] [CrossRef]
- Wang, C.-H.; Leung, C.-H.; Liu, S.-C.; Chung, C.-H. Safety and effectiveness of rosiglitazone in type 2 diabetes patients with nonalcoholic Fatty liver disease. J. Formos. Med. Assoc. 2006, 105, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.M.; Jones, F.J.; Shaw, J.C.; Williams, C.D.; Ward, J.A.; Harrison, S.A. Rosiglitazone versus rosiglitazone and metformin versus rosiglitazone and losartan in the treatment of nonalcoholic steatohepatitis in humans: A 12-month randomized, prospective, open- label trial. Hepatology 2011, 54, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, M.; Serfaty, L.; Cervera, P.; Capeau, J.; Ratziu, V. Hepatic molecular effects of rosiglitazone in human non-alcoholic steatohepatitis suggest long-term pro-inflammatory damage. Hepatol. Res. 2014, 44, 1241–1247. [Google Scholar] [CrossRef]
- Choung, S.; Joung, K.H.; You, B.R.; Park, S.K.; Kim, H.J.; Ku, B.J. Treatment with Lobeglitazone Attenuates Hepatic Steatosis in Diet-Induced Obese Mice. PPAR Res. 2018, 2018, 4292509. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.J.; Cho, Y.M. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J. Korean Med. Sci. 2017, 32, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Fiévet, C.; Fruchart, J.-C.; Staels, B. PPARalpha and PPARgamma dual agonists for the treatment of type 2 diabetes and the metabolic syndrome. Curr. Opin. Pharmacol. 2006, 6, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R. The first approved agent in the Glitazar’s Class: Saroglitazar. Curr. Drug Targets 2014, 15, 151–155. [Google Scholar] [CrossRef]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P. New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.R.; Giri, S.R.; Trivedi, C.; Bhoi, B.; Rath, A.; Vanage, G. Saroglitazar, a novel PPARα/γ agonist with predominant PPARα activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharmacol. Res. Perspect. 2015, 3, e00136. [Google Scholar] [CrossRef]
- Jain, M.R.; Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018, 38, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.P.; Caffrey, R.; Marioneaux, J.; Santhekadur, P.K.; Bhat, M.; Alonso, C. The PPAR α/γ Agonist Saroglitazar Improves Insulin Resistance and Steatohepatitis in a Diet Induced Animal Model of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 9330. [Google Scholar] [CrossRef] [PubMed]
- Jani, R.H.; Pai, V.; Jha, P.; Jariwala, G.; Mukhopadhyay, S.; Bhansali, A. A Multicenter, Prospective, Randomized, Double-Blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared with Placebo in Type 2 Diabetes Mellitus Patients Having Hypertriglyceridemia Not Controlled with Atorvastatin Therapy (PRESS VI). Diabetes Technol. Ther. 2014, 16, 63–71. [Google Scholar] [PubMed] [Green Version]
- Pai, V.; Paneerselvam, A.; Mukhopadhyay, S.; Bhansali, A.; Kamath, D.; Shankar, V. A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V). J. Diabetes Sci. Technol. 2014, 8, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, N.; Bhansali, S.; Kurpad, A.V.; Hawkins, M.; Sharma, A.; Kaur, S. Effect of a Dual PPAR α/γ agonist on Insulin Sensitivity in Patients of Type 2 Diabetes with Hypertriglyceridemia- Randomized double-blind placebo-controlled trial. Sci. Rep. 2019, 9, 19017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A. An Observational Study of Reduction in Glycemic Parameters and Liver Stiffness by Saroglitazar 4 mg in Patients with Type 2 Diabetes Mellitus and Nonalcoholic Fatty Liver Disease. Cureus 2020, 12, e9065. [Google Scholar] [CrossRef] [PubMed]
- Goyal, O.; Nohria, S.; Goyal, P.; Kaur, J.; Sharma, S.; Sood, A. Saroglitazar in patients with non-alcoholic fatty liver disease and diabetic dyslipidemia: A prospective, observational, real world study. Sci. Rep. 2020, 10, 21117. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Idowu, M.O.; Parmar, D.; Borg, B.B.; Denham, D.; Loo, N.M. A Phase 2 Double Blinded, Randomized Controlled Trial of Saroglitazar in Patients with Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2021, 19, 2670–2672. [Google Scholar] [CrossRef]
- Rubin, C.J.; Viraswami-Appanna, K.; Fiedorek, F.T. Efficacy and safety of muraglitazar: A double-blind, 24-week, dose-ranging study in patients with type 2 diabetes. Diab. Vasc. Dis. Res. 2009, 6, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Westerouen Van Meeteren, M.J.; Drenth, J.P.H.; Tjwa, E.T.T.L. Elafibranor: A potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Exp. Opin. Investig. Drugs 2020, 29, 117–123. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Cami, B.; De Boe, V. Human hepatic in vitro models reveal distinct anti-NASH potencies of PPAR agonists. Cell Biol. Toxicol. 2021, 37, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zaïr, Y.; Sauvinet, V.; Noël, B. Dual peroxisome proliferator-activated receptor α/δ agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenth, J.P.H.; Schattenberg, J.M. The nonalcoholic steatohepatitis (NASH) drug development graveyard: Established hurdles and planning for future success. Exp. Opin. Investig. Drugs 2020, 29, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Vuppalanchi, R.; Noureddin, M.; Alkhouri, N.; Sanyal, A.J. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) α/γ/δ Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, G.; Luccarini, J.; Poekes, L.; Faye, P.; Kupkowski, F.; Adarbes, V. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 2017, 1, 524–537. [Google Scholar] [CrossRef]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andrés-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernández-Iglesias, A. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2021, 74, 1188–1199. [Google Scholar] [CrossRef]
- Francque, S.; Bedossa, P.; Abdelmalek, M.F.; Anstee, Q.; Bugianesi, E.; Ratziu, V. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study. Contemp. Clin. Trials 2020, 98, 106170. [Google Scholar]
- Majzoub, A.M.; Nayfeh, T.; Barnard, A.; Munaganuru, N.; Dave, S.; Singh, S. Systematic review with network meta-analysis: Comparative efficacy of pharmacologic therapies for fibrosis improvement and resolution of NASH. Aliment. Pharm. Ther. 2021, 54, 880–889. [Google Scholar] [CrossRef]
- American Diabetes Association. Standards of Medical Care in Diabetes—2021. Diabetes Care. Available online: https://care.diabetesjournals.org/content/44/Supplement_1,/content/44/Supplement_1 (accessed on 25 October 2021).
- Balas, B.; Belfort, R.; Harrison, S.A.; Darland, C.; Finch, J.; Schenker, S. Pioglitazone treatment increases whole body fat but not total body water in patients with non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Jensen, M.D.; McCann, F.; Mukhopadhyay, D.; Joyner, M.J.; Rizza, R.A. Effects of pioglitazone versus glipizide on body fat distribution, body water content, and hemodynamics in type 2 diabetes. Diabetes Care 2006, 29, 510–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, Y.; Mahankali, A.; Wajcberg, E.; Bajaj, M.; Mandarino, L.J.; DeFronzo, R.A. Effect of pioglitazone on circulating adipocytokine levels and insulin sensitivity in type 2 diabetic patients. J. Clin. Endocrinol. Metab. 2004, 89, 4312–4319. [Google Scholar] [CrossRef] [PubMed]
- Harvey, I.; Boudreau, A.; Stephens, J.M. Adipose tissue in health and disease. Open Biol. 2020, 10, 200291. [Google Scholar] [CrossRef]
- White, U.; Fitch, M.D.; Beyl, R.A.; Hellerstein, M.K.; Ravussin, E. Adipose depot-specific effects of 16 weeks of pioglitazone on in vivo adipogenesis in women with obesity: A randomised controlled trial. Diabetologia 2021, 64, 159–167. [Google Scholar] [CrossRef]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef] [Green Version]
- Kernan, W.N.; Viscoli, C.M.; Furie, K.L.; Young, L.H.; Inzucchi, S.E.; Gorman, M. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2016, 374, 1321–1331. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef] [Green Version]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [Green Version]
- Grundy, S.M. Small LDL, atherogenic dyslipidemia, and the metabolic syndrome. Circulation 1997, 95, 1–4. [Google Scholar] [CrossRef]
- Staels, B.; Maes, M.; Zambon, A. Fibrates and future PPARalpha agonists in the treatment of cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 542–553. [Google Scholar] [CrossRef]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet Lond. Engl. 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- ACCORD Study Group; Ginsberg, H.N.; Elam, M.B.; Lovato, L.C.; Crouse, J.R.; Leiter, L.A. Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 2010, 362, 1563–1574. [Google Scholar] [PubMed]
- Fruchart, J.-C.; Hermans, M.P.; Fruchart-Najib, J.; Kodama, T. Selective Peroxisome Proliferator-Activated Receptor Alpha Modulators (SPPARMα) in the Metabolic Syndrome: Is Pemafibrate Light at the End of the Tunnel? Curr. Atheroscler. Rep. 2021, 23, 3. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T. Efficacy and safety of pemafibrate in people with type 2 diabetes and elevated triglyceride levels: 52-week data from the PROVIDE study. Diabetes Obes. Metab. 2019, 21, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Arai, H.; Yokote, K.; Araki, E.; Matsushita, M.; Nojima, T. Efficacy and Safety of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα): Pooled Analysis of Phase 2 and 3 Studies in Dyslipidemic Patients with or without Statin Combination. Int. J. Mol. Sci. 2019, 20, 5537. [Google Scholar] [CrossRef] [Green Version]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S. Efficacy and Safety of Pemafibrate Versus Fenofibrate in Patients with High Triglyceride and Low HDL Cholesterol Levels: A Multicenter, Placebo-Controlled, Double-Blind, Randomized Trial. J. Atheroscler. Thromb. 2018, 25, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Nicholls, S.J.; Wolski, K.; Nesto, R.; Kupfer, S.; Perez, A. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: The PERISCOPE randomized controlled trial. JAMA 2008, 299, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, T.; Meyer, P.M.; Feinstein, S.B.; Davidson, M.H.; Kondos, G.T.; D’Agostino, R.B. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: A randomized trial. JAMA 2006, 296, 2572–2581. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Tuzcu, E.M.; Wolski, K.; Bayturan, O.; Lavoie, A.; Uno, K. Lowering the triglyceride/high-density lipoprotein cholesterol ratio is associated with the beneficial impact of pioglitazone on progression of coronary atherosclerosis in diabetic patients: Insights from the PERISCOPE (Pioglitazone Effect on Regression of Intravascular Sonographic Coronary Obstruction Prospective Evaluation) study. J. Am. Coll. Cardiol. 2011, 57, 153–159. [Google Scholar]
- Davidson, M.; Meyer, P.M.; Haffner, S.; Feinstein, S.; D’Agostino, R.; Kondos, G.T. Increased high-density lipoprotein cholesterol predicts the pioglitazone-mediated reduction of carotid intima-media thickness progression in patients with type 2 diabetes mellitus. Circulation 2008, 117, 2123–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Majumder, A.; Ray, S. Observational study of effects of Saroglitazar on glycaemic and lipid parameters on Indian patients with type 2 diabetes. Sci. Rep. 2015, 5, 7706. [Google Scholar] [CrossRef] [Green Version]
- Dutta, D.; Bhattacharya, S.; Surana, V.; Aggarwal, S.; Singla, R.; Khandelwal, D. Efficacy and safety of saroglitazar in managing hypertriglyceridemia in type-2 diabetes: A meta-analysis. Diabetes Metab. Syndr. 2020, 14, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Krishnappa, M.; Patil, K.; Parmar, K.; Trivedi, P.; Mody, N.; Shah, C. Effect of saroglitazar 2 mg and 4 mg on glycemic control, lipid profile and cardiovascular disease risk in patients with type 2 diabetes mellitus: A 56-week, randomized, double blind, phase 3 study (PRESS XII study). Cardiovasc. Diabetol. 2020, 19, 93. [Google Scholar] [CrossRef]
- Choi, Y.-J.; Roberts, B.K.; Wang, X.; Geaney, J.C.; Naim, S.; Wojnoonski, K. Effects of the PPAR-δ agonist MBX-8025 on atherogenic dyslipidemia. Atherosclerosis 2012, 220, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Zaïr, Y.; Staels, B.; Bruckert, E. Effects of the New Dual PPARα/δ Agonist GFT505 on Lipid and Glucose Homeostasis in Abdominally Obese Patients with Combined Dyslipidemia or Impaired Glucose Metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [Green Version]
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef]
- Bernardi, M.; Caraceni, P. Novel perspectives in the management of decompensated cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 753–764. [Google Scholar] [CrossRef]
- Torres-Peña, J.D.; Martín-Piedra, L.; Fuentes-Jiménez, F. Statins in Non-alcoholic Steatohepatitis. Front. Cardiovasc. Med. 2021, 8, 777131. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.J.; Staffa, J.A.; Shatin, D.; Andrade, S.E.; Schech, S.D.; La Grenade, L. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 2004, 292, 2585–2590. [Google Scholar] [CrossRef] [Green Version]
- Davidson, M.H.; Armani, A.; McKenney, J.M.; Jacobson, T.A. Safety considerations with fibrate therapy. Am. J. Cardiol. 2007, 99, 3C–18C. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.C.T.; Delgado, V.; Borlaug, B.A.; Bax, J.J. Diabesity: The combined burden of obesity and diabetes on heart disease and the role of imaging. Nat. Rev. Cardiol. 2021, 18, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Aubin, A.; Loomba, R. The Relationship Between Type 2 Diabetes, NAFLD, and Cardiovascular Risk. Curr. Diab. Rep. 2021, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Roelstraete, B.; Khalili, H.; Hagström, H.; Ludvigsson, J.F. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: Results from a nationwide cohort. Gut 2021, 70, 1375–1382. [Google Scholar] [CrossRef]
- Dennis, B.B.; Sallam, S.; Perumpail, B.J.; Shah, N.D.; Kim, D.; Cholankeril, G. Management of Cardiometabolic Complications in Patients with Nonalcoholic Fatty Liver Disease: A Review of the Literature With Recommendations. J. Clin. Gastroenterol. 2021, 55, 747–756. [Google Scholar] [CrossRef]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef]
- Wang, D.; Liu, B.; Tao, W.; Hao, Z.; Liu, M. Fibrates for secondary prevention of cardiovascular disease and stroke. Cochrane Database Syst Rev. 2015, 2015, CD009580. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Paynter, N.P.; Everett, B.M.; Glynn, R.J.; Amarenco, P.; Elam, M. Rationale and design of the Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) study. Am. Heart J. 2018, 206, 80–93. [Google Scholar] [CrossRef]
- van der Meer, R.W.; Rijzewijk, L.J.; de Jong, H.W.A.M.; Lamb, H.J.; Lubberink, M.; Romijn, J.A. Pioglitazone improves cardiac function and alters myocardial substrate metabolism without affecting cardiac triglyceride accumulation and high-energy phosphate metabolism in patients with well-controlled type 2 diabetes mellitus. Circulation 2009, 119, 2069–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, G.D.; Solis-Herrera, C.; Molina-Wilkins, M.; Martinez, S.; Merovci, A.; Cersosimo, E. Pioglitazone Improves Left Ventricular Diastolic Function in Subjects with Diabetes. Diabetes Care 2017, 40, 1530–1536. [Google Scholar] [CrossRef] [Green Version]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.A.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial in macroVascular Events): A randomised controlled trial. Lancet Lond. Engl. 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Wolski, K.; Nicholls, S.J.; Nissen, S.E. Pioglitazone and Risk of Cardiovascular Events in Patients with Type 2 Diabetes Mellitus: A Meta-analysis of Randomized Trials. JAMA 2007, 298, 1180. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.D.; Viscoli, C.M.; Inzucchi, S.E.; Dearborn-Tomazos, J.; Ford, G.A.; Gorman, M. Pioglitazone Therapy in Patients with Stroke and Prediabetes: A Post Hoc Analysis of the IRIS Randomized Clinical Trial. JAMA Neurol. 2019, 76, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Lago, R.M.; Singh, P.P.; Nesto, R.W. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: A meta-analysis of randomised clinical trials. Lancet Lond. Engl. 2007, 370, 1129–1136. [Google Scholar] [CrossRef]
- Young, L.H.; Viscoli, C.M.; Schwartz, G.G.; Inzucchi, S.E.; Curtis, J.P.; Gorman, M.J. Heart Failure After Ischemic Stroke or Transient Ischemic Attack in Insulin-Resistant Patients Without Diabetes Mellitus Treated with Pioglitazone. Circulation 2018, 138, 1210–1220. [Google Scholar] [CrossRef]
- Lehrke, M.; Marx, N. Diabetes Mellitus and Heart Failure. Am. J. Cardiol. 2017, 120, S37–S47. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Wolski, K. Rosiglitazone revisited: An updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch. Intern. Med. 2010, 170, 1191–1201. [Google Scholar] [CrossRef]
- Graham, D.J.; Ouellet-Hellstrom, R.; MaCurdy, T.E.; Ali, F.; Sholley, C.; Worrall, C. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly Medicare patients treated with rosiglitazone or pioglitazone. JAMA 2010, 304, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Home, P.D.; Pocock, S.J.; Beck-Nielsen, H.; Curtis, P.S.; Gomis, R.; Hanefeld, M. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): A multicentre, randomised, open-label trial. Lancet Lond. Engl. 2009, 373, 2125–2135. [Google Scholar] [CrossRef]
- Borém, L.M.A.; Neto, J.F.R.; Brandi, I.V.; Lelis, D.F.; Santos, S.H.S. The role of the angiotensin II type I receptor blocker telmisartan in the treatment of non-alcoholic fatty liver disease: A brief review. Hypertens. Res. 2018, 41, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Tomita, K.; Kawai, T.; Yokoyama, H.; Shimada, A.; Kikuchi, M. Effect of Telmisartan or Losartan for Treatment of Nonalcoholic Fatty Liver Disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY). Int. J. Endocrinol. 2013, 2013, 587140. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Mok, J.S.; Han, Y.I.; Park, T.S.; Kang, K.W.; Choi, C.S. Connectivity mapping of angiotensin-PPAR interactions involved in the amelioration of non-alcoholic steatohepatitis by Telmisartan. Sci. Rep. 2019, 9, 4003. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.-F.; Caussy, C.; Loomba, R. Combination therapy for non-alcoholic steatohepatitis: Rationale, opportunities and challenges. Gut 2020, 69, 1877–1884. [Google Scholar] [CrossRef]
- Zoeller, R.T.; Brown, T.R.; Doan, L.L.; Gore, A.C.; Skakkebaek, N.E.; Soto, A.M. Endocrine-disrupting chemicals and public health protection: A statement of principles from The Endocrine Society. Endocrinology 2012, 153, 4097–4110. [Google Scholar] [CrossRef] [PubMed]
- le Maire, A.; Bourguet, W.; Balaguer, P. A structural view of nuclear hormone receptor: Endocrine disruptor interactions. Cell Mol. Life Sci. 2010, 67, 1219–1237. [Google Scholar] [CrossRef]
- Cano, R.; Pérez, J.L.; Dávila, L.A.; Ortega, Á.; Gómez, Y.; Valero-Cedeño, N.J. Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 4807. [Google Scholar] [CrossRef]
- Amato, A.A.; Wheeler, H.B.; Blumberg, B. Obesity and endocrine-disrupting chemicals. Endocr. Connect. 2021, 10, R87–R105. [Google Scholar] [CrossRef]
- Lind, P.M.; Lind, L. Endocrine-disrupting chemicals and risk of diabetes: An evidence-based review. Diabetologia 2018, 61, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Brocker, C.N.; Yan, T.; Xie, C.; Krausz, K.W.; Xiang, R. Metabolic adaptation to intermittent fasting is independent of peroxisome proliferator-activated receptor alpha. Mol. Metab. 2018, 7, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, R.; Iuculano, F.; Pallini, G.; Fargion, S.; Fracanzani, A.L. Nutrients, Genetic Factors, and Their Interaction in Non-Alcoholic Fatty Liver Disease and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 8761. [Google Scholar] [CrossRef]
- Wu, L.; Li, J.; Feng, J.; Ji, J.; Yu, Q.; Li, Y. Crosstalk between PPARs and gut microbiota in NAFLD. Biomed. Pharm. Biomed. Pharm. 2021, 136, 111255. [Google Scholar] [CrossRef] [PubMed]
- Lemberger, T.; Saladin, R.; Vázquez, M.; Assimacopoulos, F.; Staels, B.; Desvergne, B. Expression of the peroxisome proliferator-activated receptor alpha gene is stimulated by stress and follows a diurnal rhythm. J. Biol. Chem. 1996, 271, 1764–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saran, A.R.; Dave, S.; Zarrinpar, A. Circadian Rhythms in the Pathogenesis and Treatment of Fatty Liver Disease. Gastroenterology 2020, 158, 1948–1966.e1. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Nascimbeni, F.; Baldelli, E.; Marrazzo, A.; Romagnoli, D.; Lonardo, A. NAFLD as a Sexual Dimorphic Disease: Role of Gender and Reproductive Status in the Development and Progression of Nonalcoholic Fatty Liver Disease and Inherent Cardiovascular Risk. Adv. Ther. 2017, 34, 1291–1326. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F. Epidemiology of Gender Differences in Diabetes and Obesity. Adv. Exp. Med. Biol. 2017, 1043, 3–8. [Google Scholar]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.-F.; Montagner, A. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020, 63, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Burra, P.; Bizzaro, D.; Gonta, A.; Shalaby, S.; Gambato, M.; Morelli, M.C. Clinical impact of sexual dimorphism in non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Liver Int. 2021, 41, 1713–1733. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Suzuki, A. Sexual Dimorphism of NAFLD in Adults. Focus on Clinical Aspects and Implications for Practice and Translational Research. J. Clin. Med. 2020, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Suzuki, A. Nonalcoholic fatty liver disease: Does sex matter? Hepatobiliary Surg. Nutr. 2019, 8, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F.; Bairey Merz, N.; Barnes, P.J.; Brinton, R.D.; Carrero, J.-J.; DeMeo, D.L. Sex and gender: Modifiers of health, disease, and medicine. Lancet Lond. Engl. 2020, 396, 565–582. [Google Scholar] [CrossRef]
- Lefebvre, P.; Staels, B. Hepatic sexual dimorphism—Implications for non-alcoholic fatty liver disease. Nat. Rev. Endocrinol. 2021, 17, 662–670. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A.; Mitchell, J.R.; Mitchell, S.J. Sex differences in the response to dietary restriction in rodents. Curr. Opin. Physiol. 2018, 6, 28–34. [Google Scholar] [CrossRef]
- Williams, R.L.; Wood, L.G.; Collins, C.E.; Callister, R. Effectiveness of weight loss interventions—Is there a difference between men and women: A systematic review. Obes. Rev. 2015, 16, 171–186. [Google Scholar] [CrossRef]
- Wang, C.; Xu, Y. Mechanisms for Sex Differences in Energy Homeostasis. J. Mol. Endocrinol. 2019, 62, R129–R143. [Google Scholar] [CrossRef] [Green Version]
- Della Torre, S. Non-alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 2020, 11, 572490. [Google Scholar] [CrossRef]
- d’Emden, M.C.; Jenkins, A.J.; Li, L.; Zannino, D.; Mann, K.P.; Best, J.D. Favourable effects of fenofibrate on lipids and cardiovascular disease in women with type 2 diabetes: Results from the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetologia 2014, 57, 2296–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvais-Jarvis, F. Gender differences in glucose homeostasis and diabetes. Physiol. Behav. 2018, 187, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Foryst-Ludwig, A.; Clemenz, M.; Hohmann, S.; Hartge, M.; Sprang, C.; Frost, N. Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative cross-talk with PPARgamma. PLoS Genet. 2008, 4, e1000108. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.Z.; Usher, M.G.; Foley, E.L., IV; Milstone, D.S.; Brosius, F.C., III; Mortensen, R.M. Sex dimorphic actions of rosiglitazone in generalised peroxisome proliferator-activated receptor-gamma (PPAR-gamma)-deficient mice. Diabetologia 2010, 53, 1493–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Drug Name; Target Pharmaceutical Company | Reference, Country | Study Type; Treatment Duration | Participants | Intervention | Results |
|---|---|---|---|---|---|
| Pemafibrate; PPARα Kowa Pharmaceutical | Nakajima et al., 2021 [61], Japan | RCT; 72 weeks | Adults w/NAFLD defined by liver fat content ≥ 10% (MRI-PDFF), liver stiffness ≥ 2.5 kPa (MRE), ALT > 40 U/L for men, >30 U/L for women Exclusion: poorly controlled T2DM (HbA1c ≥ 8%) N = 118 Female: 50 (42%) T2DM: 43 (36%) | Arm (1): Pemafibrate 0.2 mg/day Arm (2): Placebo | Liver-related outcomes ΔIHTG by MRI-PDFF (%): (1): −5.3 (2): −4.2 ΔLSM by MRE (%): (1): −7.3 ab (2): −1.1 Significant reductions of ALT, γ-GT, and ALP in (1). No significant changes in AST. Metabolic outcomes Significant reduction of TC, LDL-C, non-HDL-C, and TG but also of HDL-C (1). |
| Yokote et al., 2021 [62], Japan | Pooled analysis of 6 RCTs; 12 weeks | Adult patients w/hypertriglyceridemia N = 1253 Female: 184 (15%) NAFLD: 534 (43%) T2DM: 449 (36%) | Arm (1): Pemafibrate 0.1 mg/day Arm (2): Pemafibrate 0.2 mg/day Arm (3): Pemafibrate 0.4 mg/day Arm (4): Placebo | Liver-related outcomes Non-significant reductions of AST (−45.5 to −58.1 U/L (1–3) vs. −33.3 U/L (4)). Dose-dependent reductions of ALT, significant for (2) and (3) (−58.5 and −67.2 U/L vs. −18.4 U/L (4)). Significant reductions in γ-GT (−61.1 to −80.6 U/L (1–3) vs. −10.9 U/L (4)). Significant reductions in ALP (1–3); non-significant reductions in bilirubin. Metabolic outcomes Significant reductions of fasting plasma glucose, fasting serum insulin, and HOMA-IR (1–3); no change in HbA1c. Significant reductions of TG and increases of HDL-C (1–3). | |
| Pioglitazone and rosiglitaone; PPARγ | Mantovani et al., 2020 [63], international (USA, Europe, and Asia) | Systematic review of 8 RCTs (pioglitazone 6 and rosiglitazone (2)); 4 to 36 months | Adults w/NAFLD and thiazolidinedione treatment for NAFLD/NASH N = 828 Female: 43% T2DM: 15% | (1): Rosiglitazone 8 mg/day (2): Pioglitazone 30 to 45 mg/day | Liver-related outcomes Significant improvements of liver fat content, NASH, and serum ALT/AST levels (1–2). No significant change in fibrosis stage compared to control in all RCTs, except one (1). |
| Musso et al., 2017 [19], international (USA, Europe, and Asia) | Meta-analysis of 8 RCTs (pioglitazone 5 and rosiglitazone (3)); 6 to 24 months | Adults w/NAFLD defined by radiological or histological evidence of steatosis N = 516 (in main analysis of primary outcome; N = 698 participants overall) Female: 333 (48%) T2DM: 142 (20%) | (1): Rosiglitazone 4 to 8 mg/day (2): Pioglitazone 30 to 45 mg/day | Liver-related outcomes Improvement of advanced fibrosis (F3-4 to F0-2; ≥2 stages improvement) in all participants (OR, 95% CI): (1): 1.30, 0.23–7.20 (2): 4.53, 1.52–13.52 Overall: 3.15, 1.25–7.93 Improvement of advanced fibrosis (F3-4 to F0-2; ≥2 stages improvement) in participants with advanced fibrosis (OR, 95% CI): (1): 1.84, 0.29–11.66 (2): 10.17, 2.83–36.54 Overall: 5.84, 2.04–16.71 Improvement of ≥1 fibrosis stage in all participants (OR, 95% CI): (1): 1.18, 0.43–3.25 (2): 4.53, 1.77, 1.15–2.72 Overall: 1.66, 1.12–2.47 NASH resolution in participants with NASH (OR, 95% CI): (1): 2.14, 0.94–4.86 (2): 3.65, 2.32–5.74 Overall: 3.22, 2.17–4.79 | |
| Pioglitazone; PPARγ | Bril et al., 2019 [64], USA | RCT; 18 months | Adults w/NASH in liver histology and T2DM Exclusion: T1DM N = 105 Female: 12 (11%) | Arm (1): Pioglitazone 45 mg/day plus vitamin E 400 IU b.i.d. Arm (2) Vitamin E 400 IU b.i.d. Arm (3): Placebo | Liver-related outcomes Reduction in NAS ≥2 points w/o worsening of fibrosis (n%): (1): 54 b (Δ 35 [14 to 56]) (2): 31 (Δ 12 [−1 to 32]) (3): 19 Reduction of NASH w/o worsening of fibrosis (% of n): (1): 43 b (Δ 31 [11 to 50]) (2): 33 b (Δ 21 [2 to 40]) (3): 12 Significant improvements in steatosis, inflammation, and ballooning (1). No significant improvements in fibrosis. Significant reduction in IHTG by 1H-MRS in (1 and 2). Metabolic outcomes Significant improvement in HbA1c (1). Modest increase in HDL-C (1). |
| Bril et al., 2019, and Cusi et al., 2018 [65,66], USA | RCT; 18 months | Adults w/NASH on liver histology, and T2DM/prediabetes Exclusion: T1DM, clinically significant renal, pulmonary, or cardiac disease N = 101 Female: 30 (30%) T2DM: 52 (51%) | Arm (1a): Pioglitazone 30 to 45 mg/day in T2DM patients Arm (1b): Pioglitazone 30 to 45 mg/day in prediabetic patients Arms (2a and b): Placebo in T2DM and prediabetic patients | Liver-related outcomes Reduction in NAS ≥2 points (≥2 different categories) w/o worsening of fibrosis (n%): (1 overall, 1a and b): 58 b, 60 ab, and 55 a (2 overall, 2a and b): 17, 16, and 29 ΔFibrosis stage (SD): (1a and b): −0.5 [0.9] b and −0.4 [0.9] (2a and b): 0.2 [1.2] and −0.2 [0.7] Significant reduction in IHTG by 1H-MRS (1a and b). Metabolic outcomes Increases in hepatic and skeletal muscle insulin sensitivity (1 and b), and in adipose tissue insulin sensitivity (1a). Significant reduction in fasting plasma insulin, and significant increases in adiponectin (1a and b), significant reduction in HbA1c (1a). Improvements in HDL-C and TG (1 and b). | |
| Saroglitazar (EVIDENCES IV); PPARα/γ Zydus Discovery | Gawrieh et al., 2021 [67], USA | RCT; 16 weeks | Adults w/NAFLD based on histology or imaging, ALT ≥ 50 U/L and BMI ≥ 25 kg/m2 Exclusion: T1DM, poorly controlled T2DM (HbA1c ≥ 9.0%) N = 106 Female: 49 (46%) T2DM: 56 (53%) | Arm (1): Saroglitazar 1 mg Arm (2): Saroglitazar 2 mg Arm (3): Saroglitazar 4 mg Arm (4): Placebo | Liver-related outcomes Significant dose-dependent ALT reductions (−25.5 to −45.8% (1–3) vs. +3.4% (4)). ALT reduction ≥25% in 64–70% (1–3) of patients compared to 18% (4). ALT reduction ≥50% in 15–52% (1–3) of patients compared to 4% (4). ALT < ULN in 6 patients (3) compared to 0 (4). Significant reductions in AST (−25.4 to 34.9% (1–3) vs. +9.8% (4)), ALP (−17.0 to 35.7% (1–3) vs. +3.3 (4)), and γ-GT (−29.4 to −45.7 (1–3) vs. +10.9 (4)). Significant reduction of steatosis by MRI-PDFF (difference −23.8% (3)) with >30% reduction in 11 patients (3) compared to 2 (4). No significant changes in CK18, LSM, or CAP. Metabolic outcomes Significant improvements in TG (1 and 3), non-significant improvements in HDL-C and LDL-C (3). Significant improvement in HOMA-IR (3), and non-significant improvements in blood glucose, HbA1c, and insulin levels (1 and 3). |
| Elafibranor (GOLDEN-505); PPARα/δ Genfit | Ratziu et al., 2016 [68], Europe and USA | RCT; 52 weeks | Adults w/NASH on liver histology N = 274 Female: 81 (33%) T2DM: 107 (39%) | Arm (1): Elafibranor 80 mg Arm (2): Elafibranor 120 mg Arm (3): Placebo | Liver-related outcomes Resolution of NASH w/o worsening of fibrosis according to current definition (OR, 95% CI): (1) 2.31, 1.02–5.24 b (2) 1.48, 0.7–3.14 Overall more pronounced effects in those with severe inflammation (NAS ≥ 4, (1): 3.52, 1.32–9.40) or presence of fibrosis (fibrosis any stage, (1): 3.75, 1.39–10.12). Improvements in γ-GT and ALP (1–2), no changes in ALT. Metabolic outcomes Significant improvements of HbA1, HOMA-IR, and fasting glucose (1) in diabetic patients, as well as improvement in plasma insulin (1–2). Dose-dependent increases in HDL-C (1–2). Decreases in LDL-C and TG (1–2). |
| Lanifibranor (NATIVE); Pan-PPAR Inventiva Pharma | Francque et al., 2021 [20], international (Europe, Canada, USA, and Australia) | RCT; 24 weeks | Adults w/NASH on liver histology Exclusion: cirrhosis (F4) N = 247 Female: 144 (58%) T2DM: 103 (2%) | Arm (1): Lanifibranor 800 mg Arm (2): Lanifibranor 1200 mg Arm (3): Placebo | Liver-related outcomes Decrease of ≥2 points from SAF activity score w/o worsening of fibrosis (RR, 95% CI): (1) 1.45, 1.00–2.10 b (2) 1.69, 1.22–2.34 b Improvement of NASH w/o worsening of fibrosis (RR, 95% CI): (1) 1.70, 1.07–2.71 b (2) 2.20, 1.49–3.26 b Improvement of fibrosis ≥1 stage w/o worsening of NASH (RR, 95% CI): (1) 1.15, 0.72–1.85 (2) 1.68, 1.15–2.46 b Significant reductions in AST, ALT, and γ-GT (1–2). Metabolic outcomes Significant improvements in HOMA-IR (−5.5 to −5.8 (1–2) vs. −1.47 (3)), insulin (−115 to −119 pmol/L (1–2) vs. −36 pmol/L (3)), HbA1c, and fasting glucose (1–2). Significant reduction of TG (1–2), no significant changes in TC, HDL-C, and LDL-C. |
| Drug Name; Target Pharmaceutical Company | Country | Trial Registration ID Trial Name | Participants (Randomization) | Intervention | Treatment Duration (Weeks) | Primary Outcome(s) Secondary Outcome(s) |
|---|---|---|---|---|---|---|
| Pioglitazone; PPARγ | South Korea | NCT03646292 | Adults w/NAFLD (diagnosed on ultrasound and other modalities) and T2DM N = 60 (1:1:1) | Arm (1): Empagliflozin 10 mg Arm (2): Pioglitazone 15 mg Arm (3): Empagliflozin 10 mg + pioglitazone 15 mg | 24 | Change in hepatic fat content by MRI-PDFF. Change in liver fibrosis by MRE, changes in lipid profiles, liver enzymes, glucose metabolism, and inflammatory biomarkers. |
| Pakistan | NCT04976283 | Adults w/NAFLD (diagnosed by FibroScan) and T2DM N = 123 (1:1:1) | Arm (1): Pioglitazone 15 mg Arm (2): Empagliflozin 5–12.5 mg Arm (3): Empagliflozin 10 mg + Pioglitazone 15 mg | 52 | Change in radiologic liver parameters. Changes in liver enzymes, liver fibrosis scores, body weight, body composition, glucose metabolism, and lipid profiles. | |
| USA | NCT04501406 | Adults w/NASH (histologically confirmed) and T2DM N = 138 (1:1) | Arm (1): Pioglitazone 15 mg Arm (2): Placebo | 72 | Proportion of patients achieving ≥2 points improvement in NAS w/o worsening of fibrosis. Resolution of NASH w/o worsening of fibrosis, improvements in SAF score and NAS, and change in fibrosis. | |
| Saroglitazar; PPARα/γ Zydus Discovery | USA | NCT03639623 EVIDENCES VIII | Adults 6 months post-transplantation for NASH N = 15 | Arm (1): Saroglitazar 4 mg | 24 | Safety (adverse events). Changes in hepatic fat, metabolic flexibility, lipid profiles, liver enzymes, glucose metabolism, pharmacokinetics, and quality of life. |
| USA | NCT05011305 | Adults w/NASH (histologically confirmed) N = 240 (1:1:1) | Arm (1): Saroglitazar 2 mg Arm (2): Saroglitazar 4 mg Arm (3): Placebo | 76 | Resolution of NASH w/o worsening of fibrosis. Improvements in fibrosis, NAS and SAF score, changes in lipid profiles, liver enzymes, glucose metabolism, and body weight. | |
| USA | NCT03617263 | Adult females w/NAFLD and PCOSN = 90 (1:1) | Arm (1): Saroglitazar 4 mg Arm (2): Placebo | 34 | Hepatic fat content by MRI-PDFF. Changes in liver enzymes, liver steatosis, liver fibrosis, BMI, body composition, glucose metabolism, pharmacokinetics, ovarian function, and free androgen index. | |
| India | NCT04193982 | Adults w/NAFLD (histologically confirmed) N = 250 (1:1:1:1) | Arm (1): Saroglitazar 4 mg Arm (2): Vitamin E 400 mg Arm (3): Saroglitazar 4 mg + vitamin E 400 mg Arm (4): Lifestyle intervention | 24 | Change in NFS. Changes in liver enzymes, lipid profiles, liver fibrosis in histology, NAS, and HbA1c. | |
| Lanifibranor; Pan-PPAR Inventiva Pharma | USA | NCT03459079 | Adults w/NAFLD and T2DM N = 44 (1:1) | Arm (1): Lanifibranor 800 mg Arm (2): Placebo | 24 | Change in IHTG by 1H-MRS. Proportion patients w/ ≥ 30% decrease in IHTG and NAFLD resolution, changes in insulin sensitivity, lipid profiles, glucose metabolism, and biomarkers of fibrosis. |
| USA | NCT04849728 NATIV3 | Adults w/NASH (histologically confirmed) and fibrosis stages 2–3 N = 2000 (1:1:1) | Arm (1): Lanifibranor 800 mg Arm (2): Lanifibranor 1200 mg Arm (3): Placebo | 72 | Resolution of NASH and improvement of fibrosis, and time to first clinical outcome event. Resolution of NASH w/o worsening of fibrosis, improvement of fibrosis w/o worsening of NASH, changes in liver enzymes, lipid profiles, glucose metabolism, and quality of life. |
| PPAR Target | Drug Name | Overweight and Obesity | Effects on Lipid Levels | Cardiovascular Comorbidities |
|---|---|---|---|---|
| PPARα | Pemafibrate | No effect | ↑ HDL-C ↓(↓) Triglycerides | Unknown; phase 3 trial (PROMINENT) ongoing |
| Fibrates | No effect | ↑ HDL-C ↓ Triglycerides | Modest decrease of cardiovascular risk in primary and secondary prevention | |
| PPARδ | Seladelpar | No effect | ↑ HDL-C ↓ LDL-C ↓ Triglycerides | No data on cardiovascular outcomes; good cardiovascular safety profile |
| PPARγ | Pioglitazone and other thiazolidinedione | Weight gain (3–7% of body weight) | ↑ HDL-C (↑) LDL-C ↓ Triglycerides | Risk reduction for several cardiovascular outcomes; causing fluid retention and edema; possibly increased cardiovascular risk with rosiglitazone |
| PPARα/γ | Saroglitazar | No effect | (↑) HDL-C (↓) LDL-C ↓ Triglycerides | No data on cardiovascular outcomes; good cardiovascular safety profile |
| PPARα/δ | Elafibranor | No effect | (↑) HDL-C ↓ Triglycerides | No data on cardiovascular outcomes; good cardiovascular safety profile |
| Pan-PPAR | Lanifibranor | Mild weight gain (3% of body weight) | ↑ HDL-C ↓ Triglycerides | No data on cardiovascular outcomes; good cardiovascular safety profile |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.-F. PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients. Int. J. Mol. Sci. 2022, 23, 4305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084305
Lange NF, Graf V, Caussy C, Dufour J-F. PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients. International Journal of Molecular Sciences. 2022; 23(8):4305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084305
Chicago/Turabian StyleLange, Naomi F., Vanessa Graf, Cyrielle Caussy, and Jean-François Dufour. 2022. "PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients" International Journal of Molecular Sciences 23, no. 8: 4305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084305