
Engineered Neutral Phosphorous Dendrimers Protect Mouse Cortical Neurons and Brain Organoids from Excitotoxic Death

 , , and
, , and 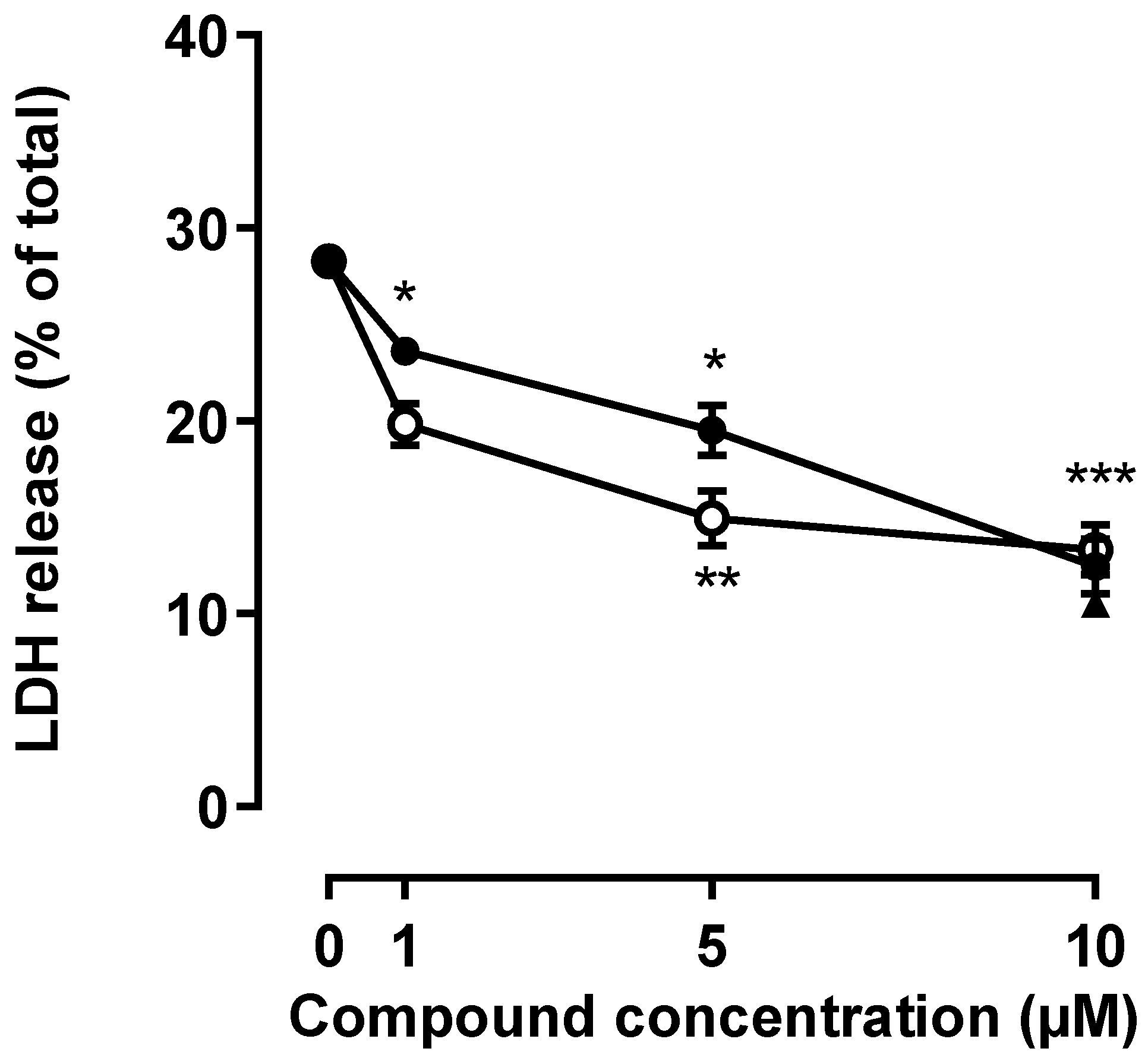{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Phosphorous Dendrimers on Excitotoxicity
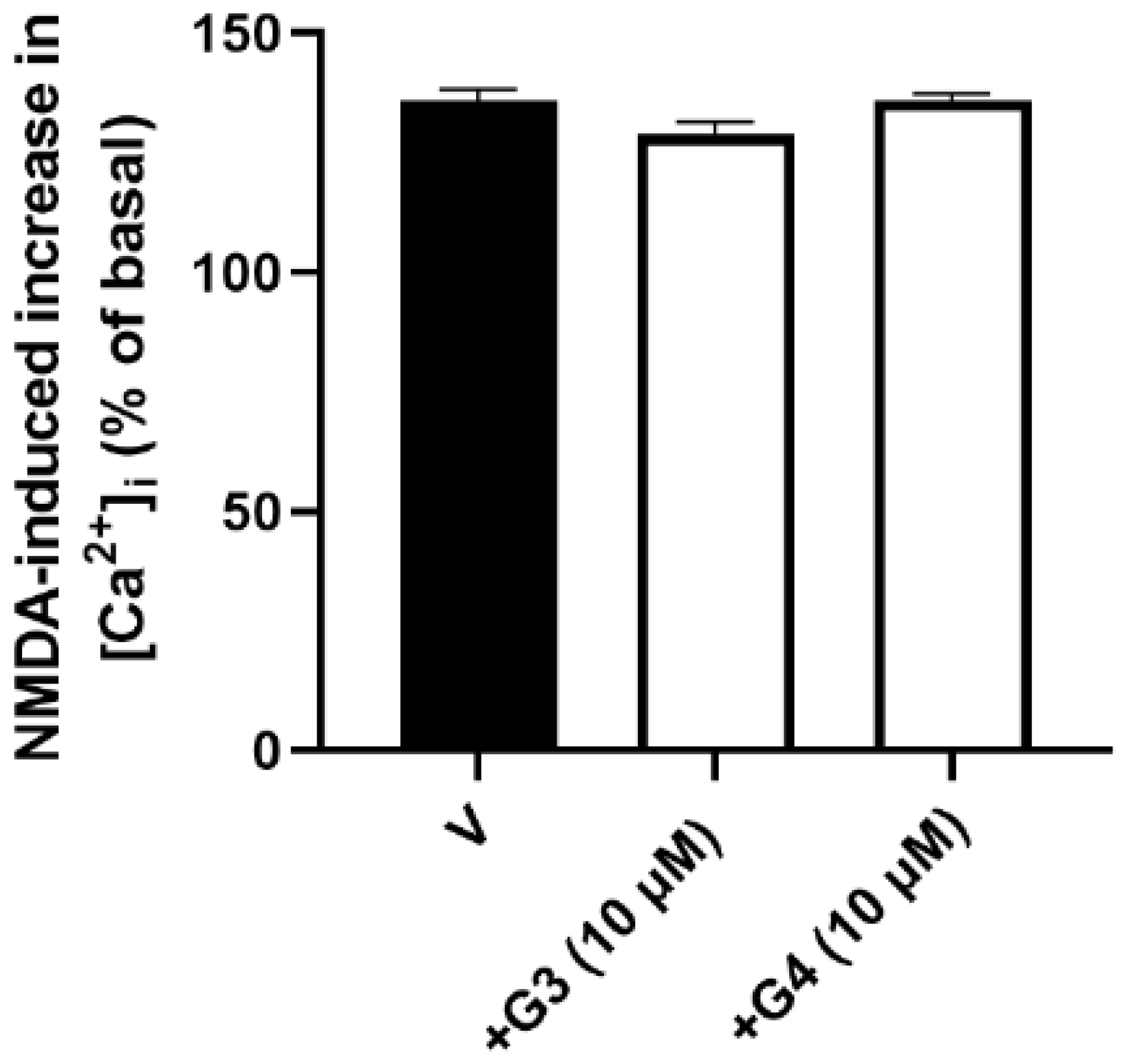
2.2. Phosphorous Dendrimers Lack Any Effect on an NMDA-Induced Increase in [Ca2+]i
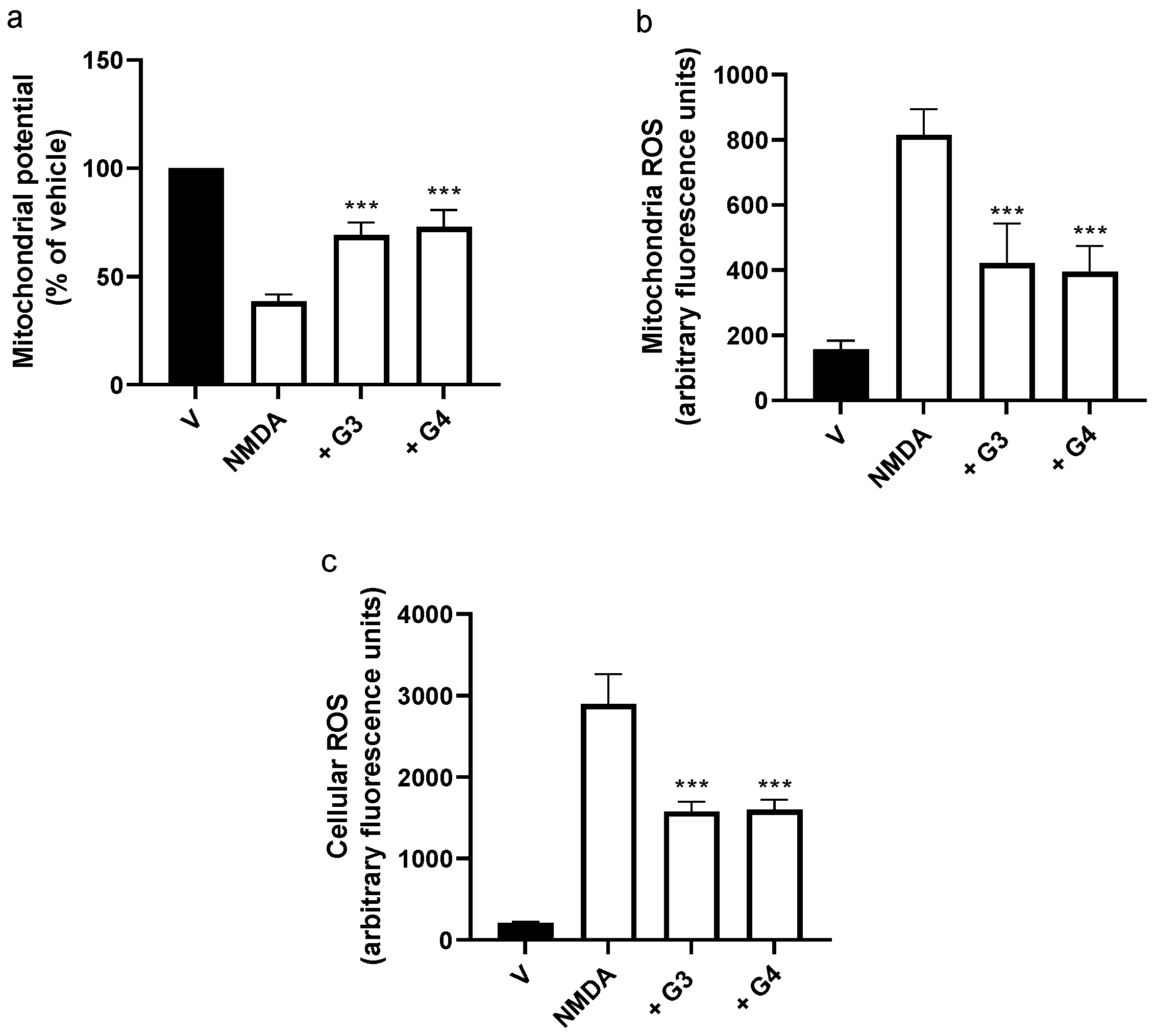
2.3. Phosphorous Dendrimers Decrease NMDA-Induced ROS Production
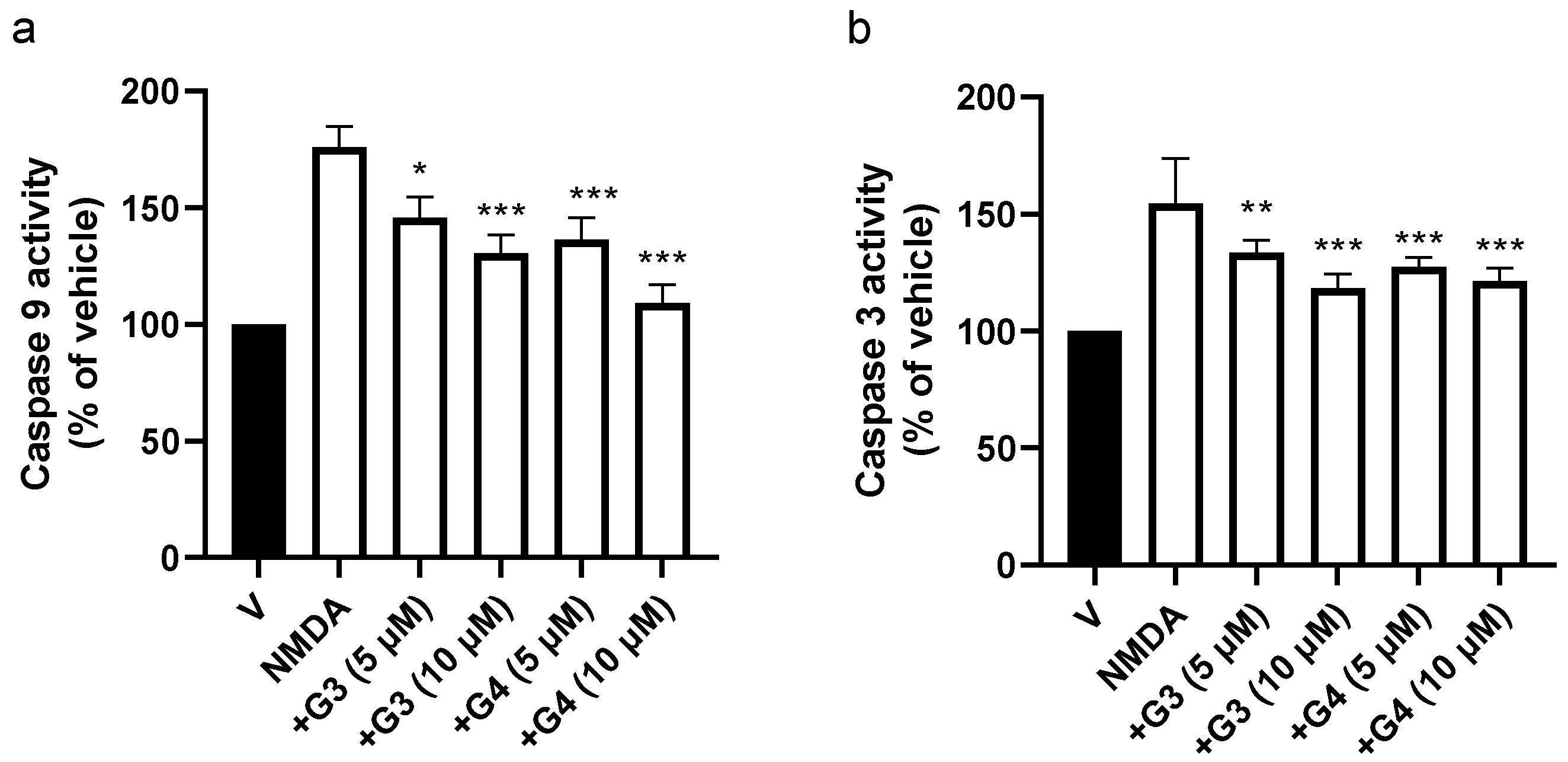
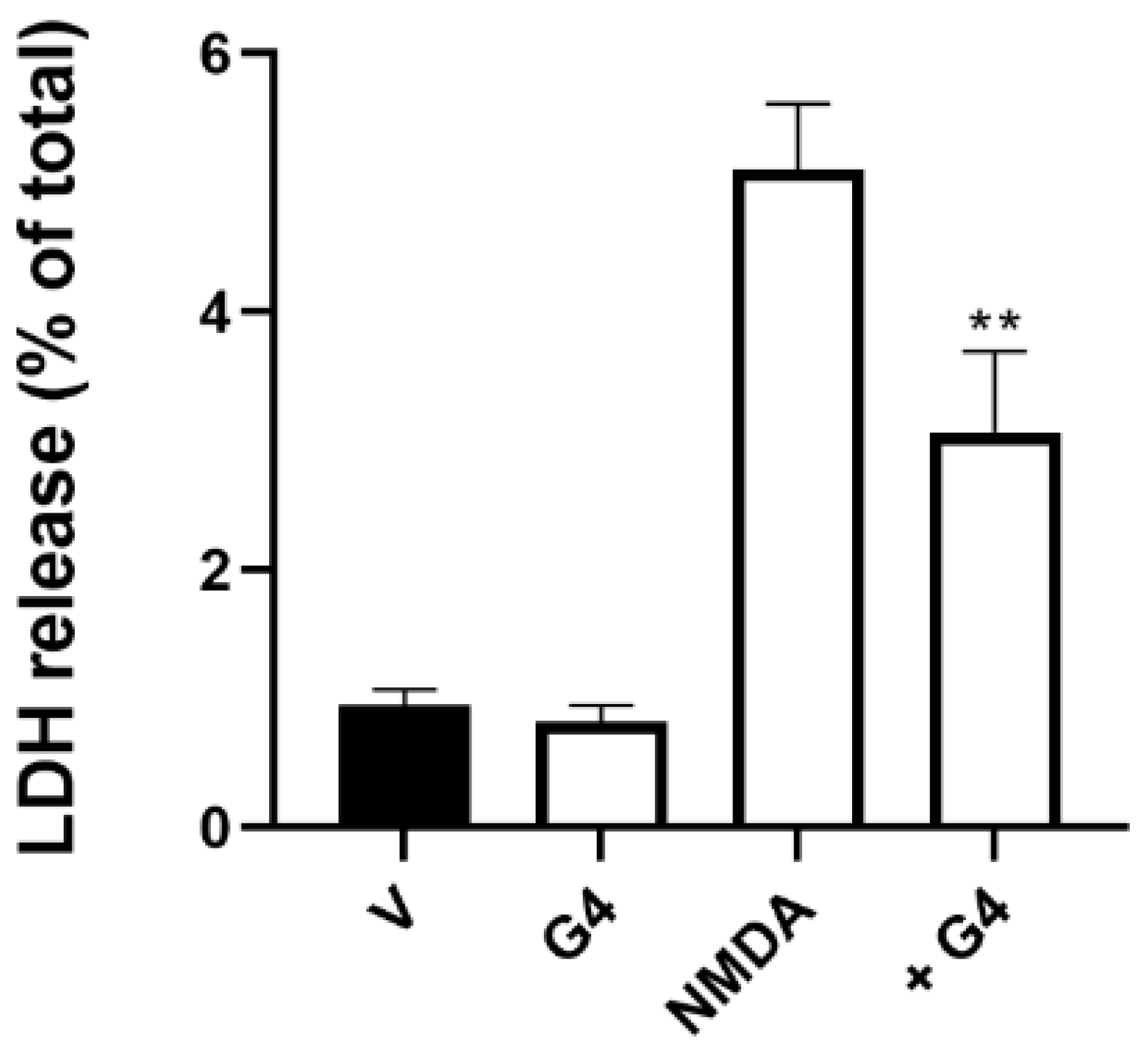
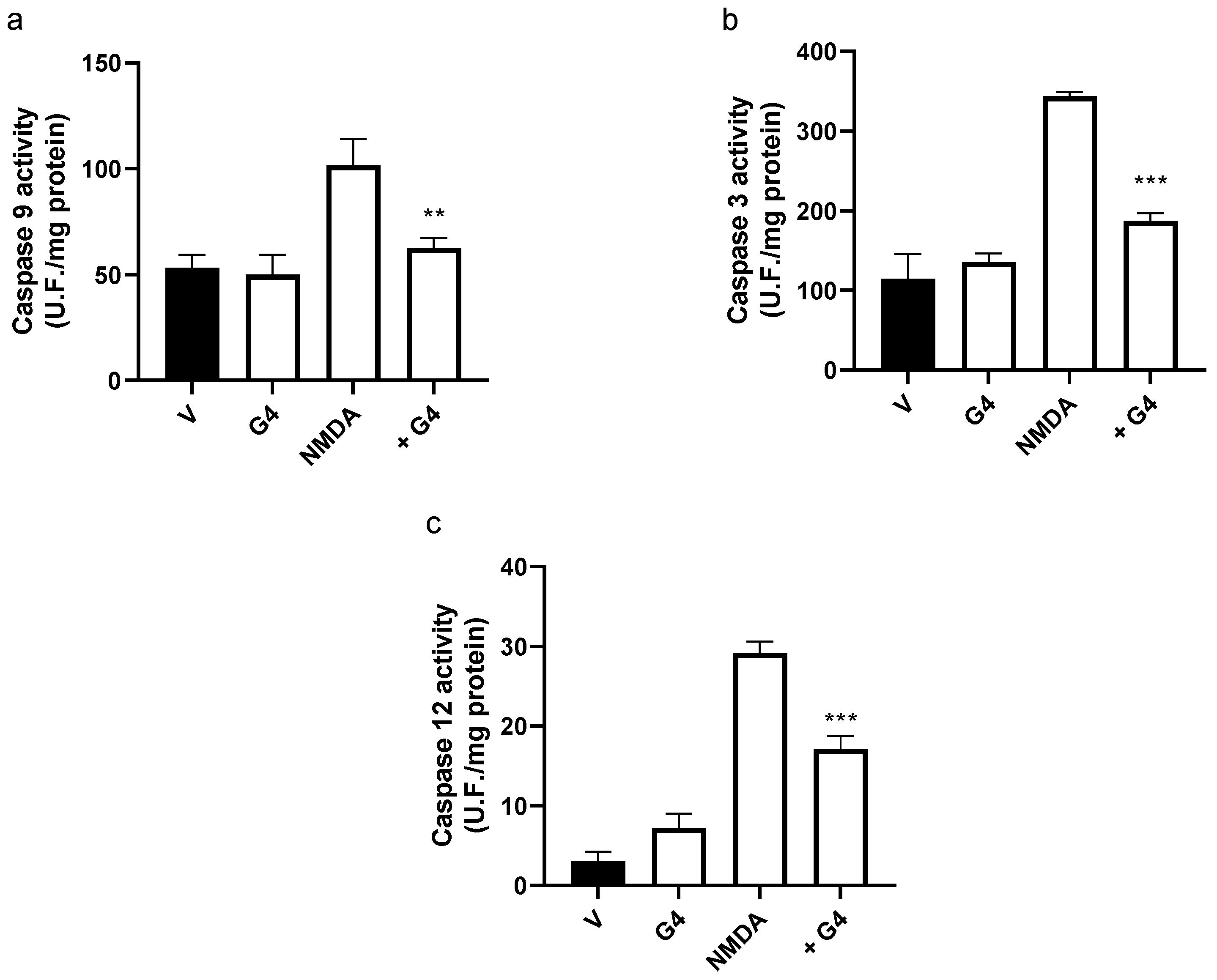
2.4. Phosphorous Dendrimers Inhibit NMDA-Induced Intrinsic Apoptotic Pathway Activation
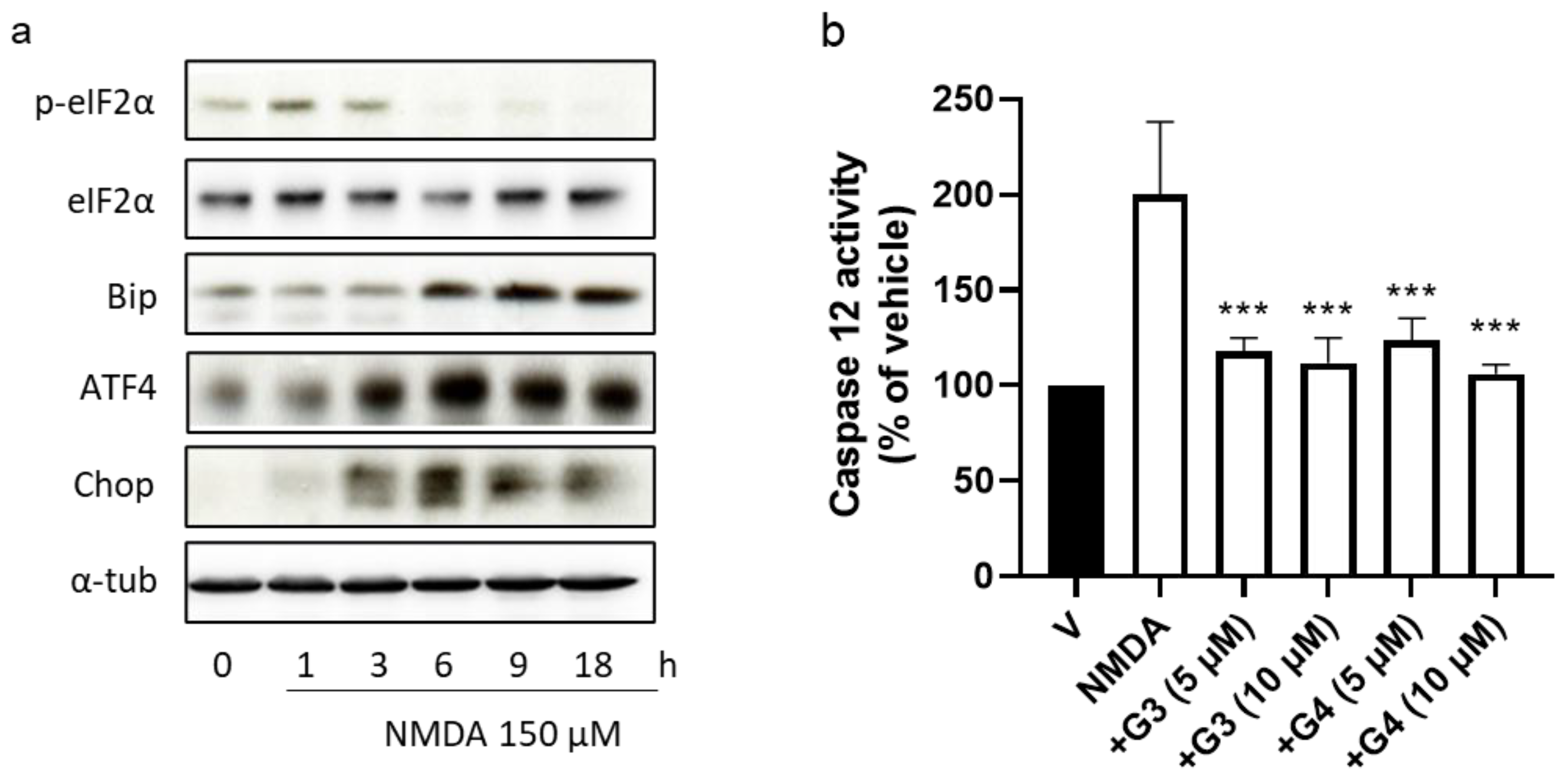
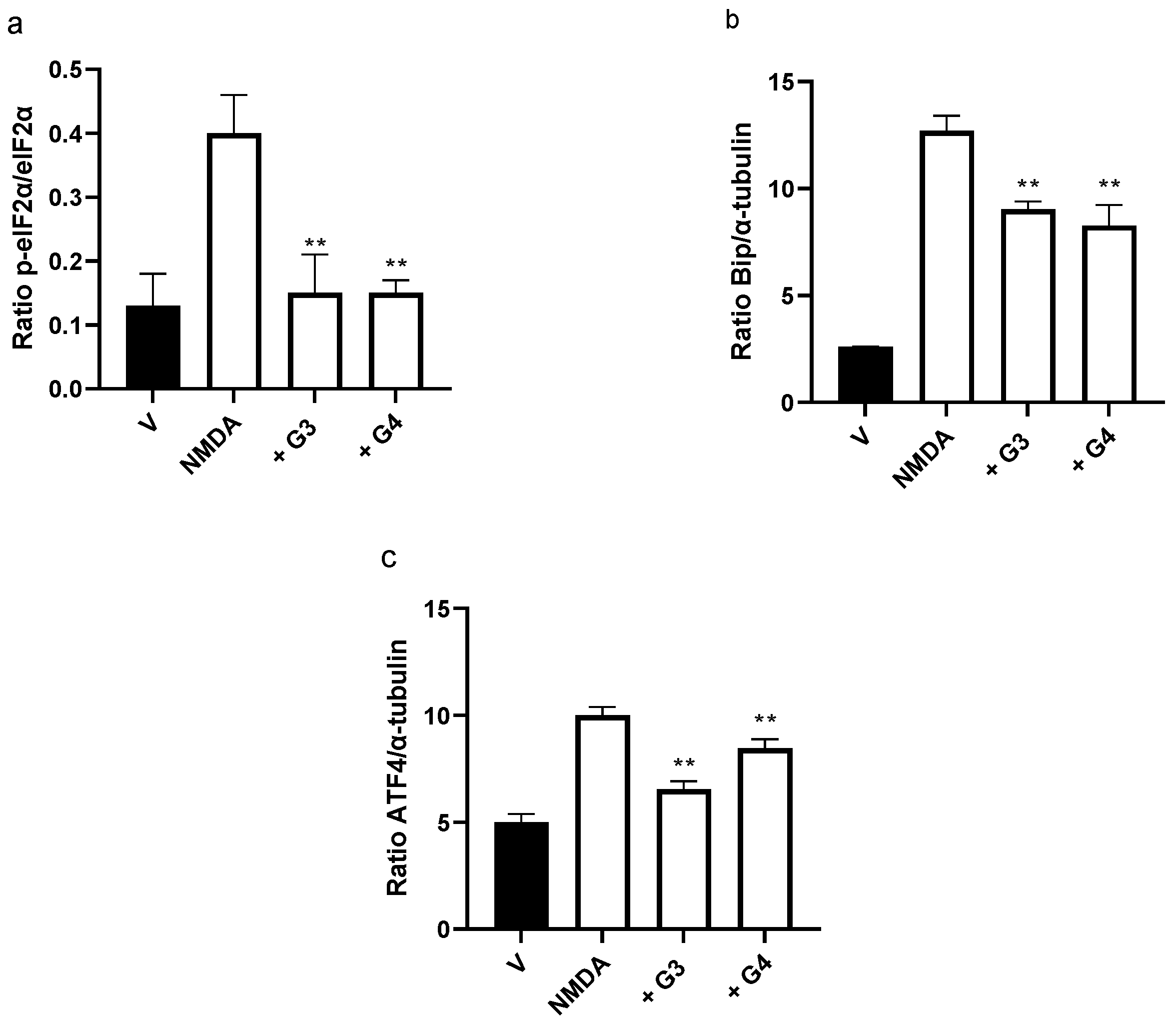
2.5. Phosphorous Dendrimers Inhibit NMDA-Induced Activation of the ER Stress Pathway
2.6. Phosphorous Dendrimers Inhibit NMDA-Induced Excitotoxicity in Brain Organoids
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.2. Animals
4.3. Cell Culture
4.4. Brain Organoids
4.5. Cellular Toxicity
4.6. Intracellular Ca2+ Measurement
4.7. ROS Generation
4.8. Mitochondrial Transmembrane Potential
4.9. Extraction of Total Lysates
4.10. Caspase Activities Determination
4.11. Western Blot Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Li, S.; Wang, X.; Chen, Q.; He, Z.; Luo, C.; Sun, J. Smart transformable nanomedicines for cancer therapy. Biomaterials 2021, 271, 120737. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiro, P.A.; Rees, D.C.; Stolzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update post COVID-19 vaccines. Bioeng. Transl. Med. 2021, 6, e10246. [Google Scholar] [CrossRef]
- Global Burden Disease Dementia 2016. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Cena, V.; Jativa, P. Nanoparticle crossing of blood-brain barrier: A road to new therapeutic approaches to central nervous system diseases. Nanomedicine 2018, 13, 1513–1516. [Google Scholar] [CrossRef] [Green Version]
- Chouhan, R.S.; Horvat, M.; Ahmed, J.; Alhokbany, N.; Alshehri, S.M.; Gandhi, S. Magnetic Nanoparticles-A Multifunctional Potential Agent for Diagnosis and Therapy. Cancers 2021, 13, 2213. [Google Scholar] [CrossRef]
- Kashapov, R.; Ibragimova, A.; Pavlov, R.; Gabdrakhmanov, D.; Kashapova, N.; Burilova, E.; Zakharova, L.; Sinyashin, O. Nanocarriers for Biomedicine: From Lipid Formulations to Inorganic and Hybrid Nanoparticles. Int. J. Mol. Sci. 2021, 22, 7055. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A New Class of Polymers–Starburst-Dendritic Macromolecules. Polym. J. 1985, 17, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Zhong, D.; Wu, H.; Wu, Y.; Li, Y.; Yang, J.; Gong, Q.; Luo, K.; Gu, Z. Redox dual-responsive dendrimeric nanoparticles for mutually synergistic chemo-photodynamic therapy to overcome drug resistance. J. Control. Release 2021, 329, 1210–1221. [Google Scholar] [CrossRef]
- Posadas, I.; Perez-Martinez, F.C.; Guerra, J.; Sanchez-Verdu, P.; Ceña, V. Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J. Neurochem. 2012, 120, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Posadas, I.; Romero-Castillo, L.; El, B.N.; Manzanares, D.; Mignani, S.; Majoral, J.P.; Ceña, V. Neutral high-generation phosphorus dendrimers inhibit macrophage-mediated inflammatory response in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E7660–E7669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solassol, J.; Crozet, C.; Perrier, V.; Leclaire, J.; Beranger, F.; Caminade, A.M.; Meunier, B.; Dormont, D.; Majoral, J.P.; Lehmann, S. Cationic phosphorus-containing dendrimers reduce prion replication both in cell culture and in mice infected with scrapie. J. Gen. Virol. 2004, 85, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Mignani, S.; Rodrigues, J.; Roy, R.; Shi, X.; Cena, V.; El Kazzouli, S.; Majoral, J.P. Exploration of biomedical dendrimer space based on in-vivo physicochemical parameters: Key factor analysis (Part 2). Drug Discov. Today 2019, 24, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Huch, M.; Koo, B.K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [Green Version]
- Corro, C.; Novellasdemunt, L.; Li, V.S.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Mansour, A.A.; Schafer, S.T.; Gage, F.H. Cellular complexity in brain organoids: Current progress and unsolved issues. Semin. Cell Dev. Biol. 2021, 111, 32–39. [Google Scholar] [CrossRef]
- Chen, H.I.; Song, H.; Ming, G.L. Applications of Human Brain Organoids to Clinical Problems. Dev. Dyn. 2019, 248, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Joffe, M.E.; Santiago, C.I.; Stansley, B.J.; Maksymetz, J.; Gogliotti, R.G.; Engers, J.L.; Nicoletti, F.; Lindsley, C.W.; Conn, P.J. Mechanisms underlying prelimbic prefrontal cortex mGlu3/mGlu5-dependent plasticity and reversal learning deficits following acute stress. Neuropharmacology 2019, 144, 19–28. [Google Scholar] [CrossRef]
- Maiolino, M.; O’Neill, N.; Lariccia, V.; Amoroso, S.; Sylantyev, S.; Angelova, P.R.; Abramov, A.Y. Inorganic Polyphosphate Regulates AMPA and NMDA Receptors and Protects Against Glutamate Excitotoxicity via Activation of P2Y Receptors. J. Neurosci. 2019, 39, 6038–6048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell Mol. Neurobiol. 2020, 41, 1413–1430. [Google Scholar] [CrossRef] [PubMed]
- Wroge, C.M.; Hogins, J.; Eisenman, L.; Mennerick, S. Synaptic NMDA receptors mediate hypoxic excitotoxic death. J. Neurosci. 2012, 32, 6732–6742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briston, T.; Hicks, A.R. Mitochondrial dysfunction and neurodegenerative proteinopathies: Mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 2018, 46, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Esteves, A.R.; Cardoso, S.M.; Oliveira, C.R. Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: Relevance to Parkinson’s disease. Neurochem. Int. 2009, 55, 341–348. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling–From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef]
- Ruiz, A.; Matute, C.; Alberdi, E. Endoplasmic reticulum Ca(2+) release through ryanodine and IP(3) receptors contributes to neuronal excitotoxicity. Cell Calcium. 2009, 46, 273–281. [Google Scholar] [CrossRef]
- Chamorro, A.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Luetjens, C.M.; Bui, N.T.; Sengpiel, B.; Munstermann, G.; Poppe, M.; Krohn, A.J.; Bauerbach, E.; Krieglstein, J.; Prehn, J.H. Delayed mitochondrial dysfunction in excitotoxic neuron death: Cytochrome c release and a secondary increase in superoxide production. J. Neurosci. 2000, 20, 5715–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Mignani, S.; Shi, X.; Ceña, V.; Shcharbin, D.; Bryszewska, M.; Majoral, J.P. In vivo therapeutic applications of phosphorus dendrimers: State of the art. Drug Discov. Today 2021, 26, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef] [PubMed]
- Zadori, D.; Veres, G.; Szalardy, L.; Klivenyi, P.; Vecsei, L. Alzheimer’s Disease: Recent Concepts on the Relation of Mitochondrial Disturbances, Excitotoxicity, Neuroinflammation, and Kynurenines. J. Alzheimers Dis. 2018, 62, 523–547. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front. Neurosci. 2020, 14, 585584. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar]
- Lindsay, A.; Hickman, D.; Srinivasan, M. A nuclear factor-kappa B inhibiting peptide suppresses innate immune receptors and gliosis in a transgenic mouse model of Alzheimer’s disease. Biomed. Pharmacother. 2021, 138, 111405. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Navarrete, L.P.; Gonzalez, A.; Gonzalez-Canacer, A.; Guzman-Martinez, L.; Cortes, N. Inflammation: A Major Target for Compounds to Control Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1199–1213. [Google Scholar] [CrossRef]
- Chinopoulos, C. Mitochondrial permeability transition pore: Back to the drawing board. Neurochem. Int. 2018, 117, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinder, A.F.; Olson, E.C.; Spitzer, N.C.; Montal, M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 1996, 16, 6125–6133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polster, B.M.; Fiskum, G. Mitochondrial mechanisms of neural cell apoptosis. J. Neurochem. 2004, 90, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, M.; Balusikova, K.; Schmiedlova, M.; Nemcova-Furstova, V.; Sramek, J.; Stancikova, J.; Zanardi, I.; Ojima, I.; Kovar, J. The role of individual caspases in cell death induction by taxanes in breast cancer cells. Cancer Cell Int. 2015, 15, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, M.; Leal-Campanario, R.; Campos-Esparza, M.R.; Sanchez-Gomez, M.V.; Alberdi, E.; Arranz, A.; Delgado-Garcia, J.M.; Gruart, A.; Matute, C. Neuroprotection by two polyphenols following excitotoxicity and experimental ischemia. Neurobiol. Dis. 2006, 23, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Shah, F.A.; Ali, T.; Tan, Z.; Alattar, A.; Ullah, N.; Khan, A.U.; Alshaman, R.; Li, S. Potent Natural Antioxidant Carveol Attenuates MCAO-Stress Induced Oxidative, Neurodegeneration by Regulating the Nrf-2 Pathway. Front. Neurosci. 2020, 14, 659. [Google Scholar] [CrossRef] [PubMed]
- Herrando-Grabulosa, M.; Gaja-Capdevila, N.; Vela, J.M.; Navarro, X. Sigma 1 receptor as a therapeutic target for amyotrophic lateral sclerosis. Br. J. Pharmacol. 2021, 178, 1336–1352. [Google Scholar] [CrossRef]
- Marchi, S.; Bittremieux, M.; Missiroli, S.; Morganti, C.; Patergnani, S.; Sbano, L.; Rimessi, A.; Kerkhofs, M.; Parys, J.B.; Bultynck, G.; et al. Endoplasmic Reticulum-Mitochondria Communication Through Ca(2+) Signaling: The Importance of Mitochondria-Associated Membranes (MAMs). Adv. Exp. Med. Biol. 2017, 997, 49–67. [Google Scholar]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef]
- Pan, C.; Prentice, H.; Price, A.L.; Wu, J.Y. Beneficial effect of taurine on hypoxia- and glutamate-induced endoplasmic reticulum stress pathways in primary neuronal culture. Amino Acids 2012, 43, 845–855. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Lee, S.; Choi, N.; Kim, H.N. Emerging Brain-Pathophysiology-Mimetic Platforms for Studying Neurodegenerative Diseases: Brain Organoids and Brains-on-a-Chip. Adv. Healthc. Mater. 2021, 10, e2002119. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Carrion, M.D.; Perez-Martinez, F.C.; Merino, S.; Sanchez-Verdu, P.; Martinez-Hernandez, J.; Lujan, R.; Ceña, V. Dendrimer-mediated siRNA delivery knocks down Beclin 1 and potentiates NMDA-mediated toxicity in rat cortical neurons. J. Neurochem. 2012, 120, 259–268. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Zhu, Y.; Qin, J. Human brain organoid-on-a-chip to model prenatal nicotine exposure. Lab Chip 2018, 18, 851–860. [Google Scholar] [CrossRef]
- Lopez-Hernandez, B.; Posadas, I.; Podlesniy, P.; Abad, M.A.; Trullas, R.; Ceña, V. HIF-1alpha is neuroprotective during the early phases of mild hypoxia in rat cortical neurons. Exp. Neurol. 2012, 233, 543–554. [Google Scholar] [CrossRef]
- Posadas, I.; Santos, P.; Blanco, A.; Muñoz-Fernandez, M.; Ceña, V. Acetaminophen induces apoptosis in rat cortical neurons. PLoS ONE 2010, 5, e15360. [Google Scholar] [CrossRef]
- Posadas, I.; Vellecco, V.; Santos, P.; Prieto-Lloret, J.; Ceña, V. Acetaminophen potentiates staurosporine-induced death in a human neuroblastoma cell line. Br. J. Pharmacol. 2007, 150, 577–585. [Google Scholar] [CrossRef]
- Liu, X.; Chen, R.; Shang, Y.; Jiao, B.; Huang, C. Superoxide radicals scavenging and xanthine oxidase inhibitory activity of magnesium lithospermate B from Salvia miltiorrhiza. J. Enzym. Inhib. Med. Chem. 2008, 24, 663–668. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posadas, I.; Romero-Castillo, L.; Ronca, R.-A.; Karpus, A.; Mignani, S.; Majoral, J.-P.; Muñoz-Fernández, M.; Ceña, V. Engineered Neutral Phosphorous Dendrimers Protect Mouse Cortical Neurons and Brain Organoids from Excitotoxic Death. Int. J. Mol. Sci. 2022, 23, 4391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084391
Posadas I, Romero-Castillo L, Ronca R-A, Karpus A, Mignani S, Majoral J-P, Muñoz-Fernández M, Ceña V. Engineered Neutral Phosphorous Dendrimers Protect Mouse Cortical Neurons and Brain Organoids from Excitotoxic Death. International Journal of Molecular Sciences. 2022; 23(8):4391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084391
Chicago/Turabian StylePosadas, Inmaculada, Laura Romero-Castillo, Rosa-Anna Ronca, Andrii Karpus, Serge Mignani, Jean-Pierre Majoral, Mariángeles Muñoz-Fernández, and Valentín Ceña. 2022. "Engineered Neutral Phosphorous Dendrimers Protect Mouse Cortical Neurons and Brain Organoids from Excitotoxic Death" International Journal of Molecular Sciences 23, no. 8: 4391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23084391