
Alchemical Design of Pharmacological Chaperones with Higher Affinity for Phenylalanine Hydroxylase

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
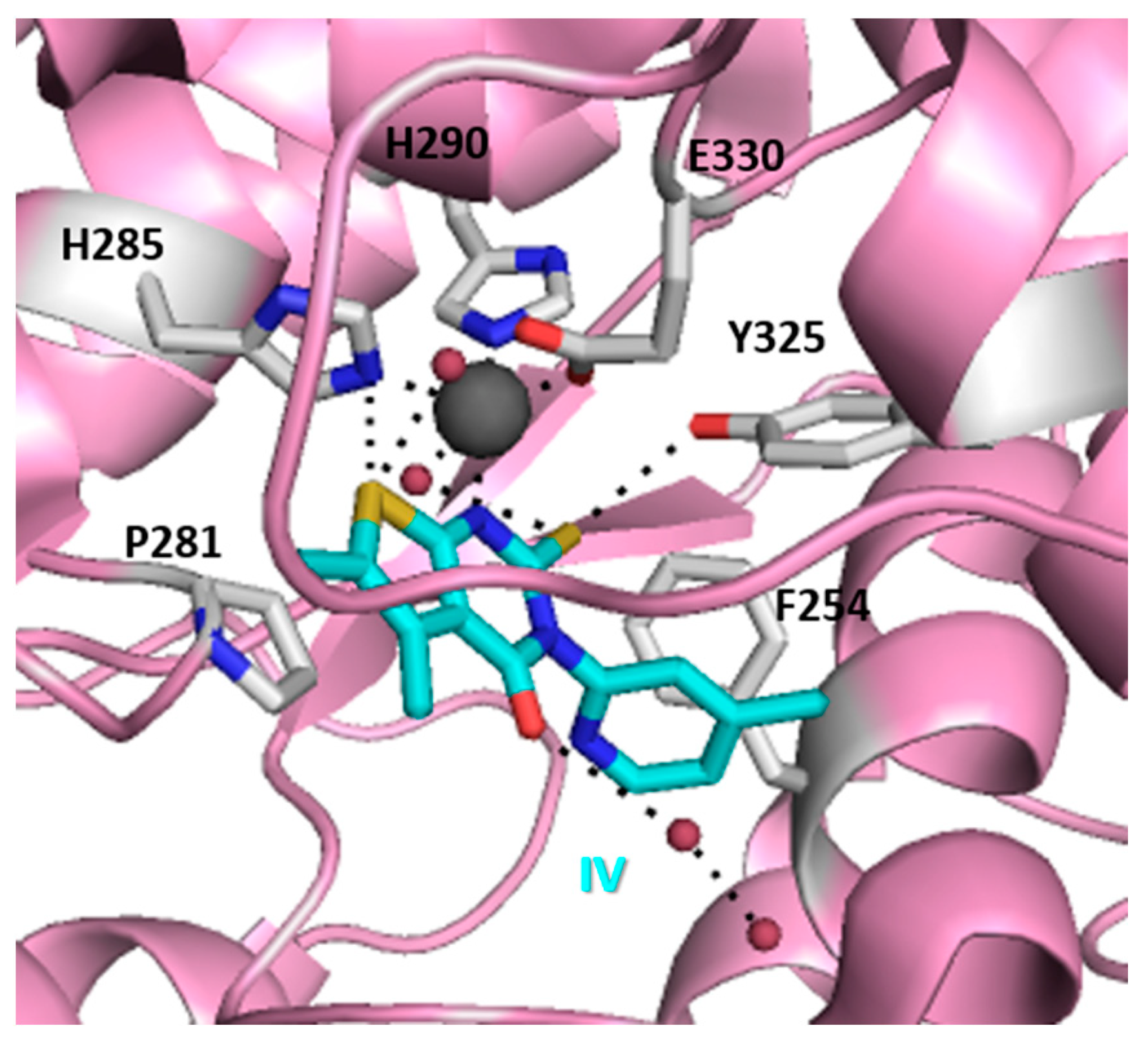
2.1. Design of Novel IVPC Analogues as Potential Pharmacological Chaperones with New Chemical Properties
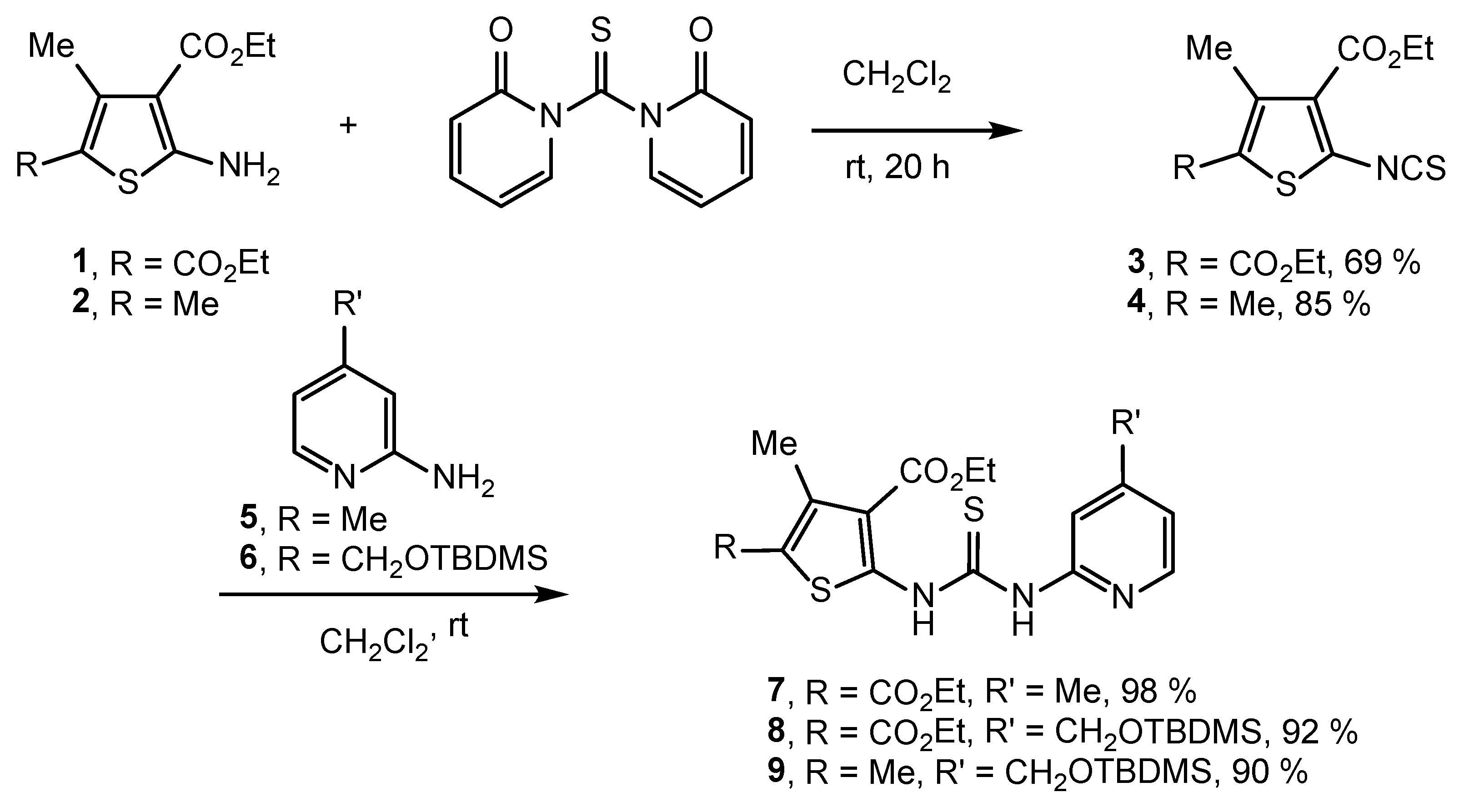
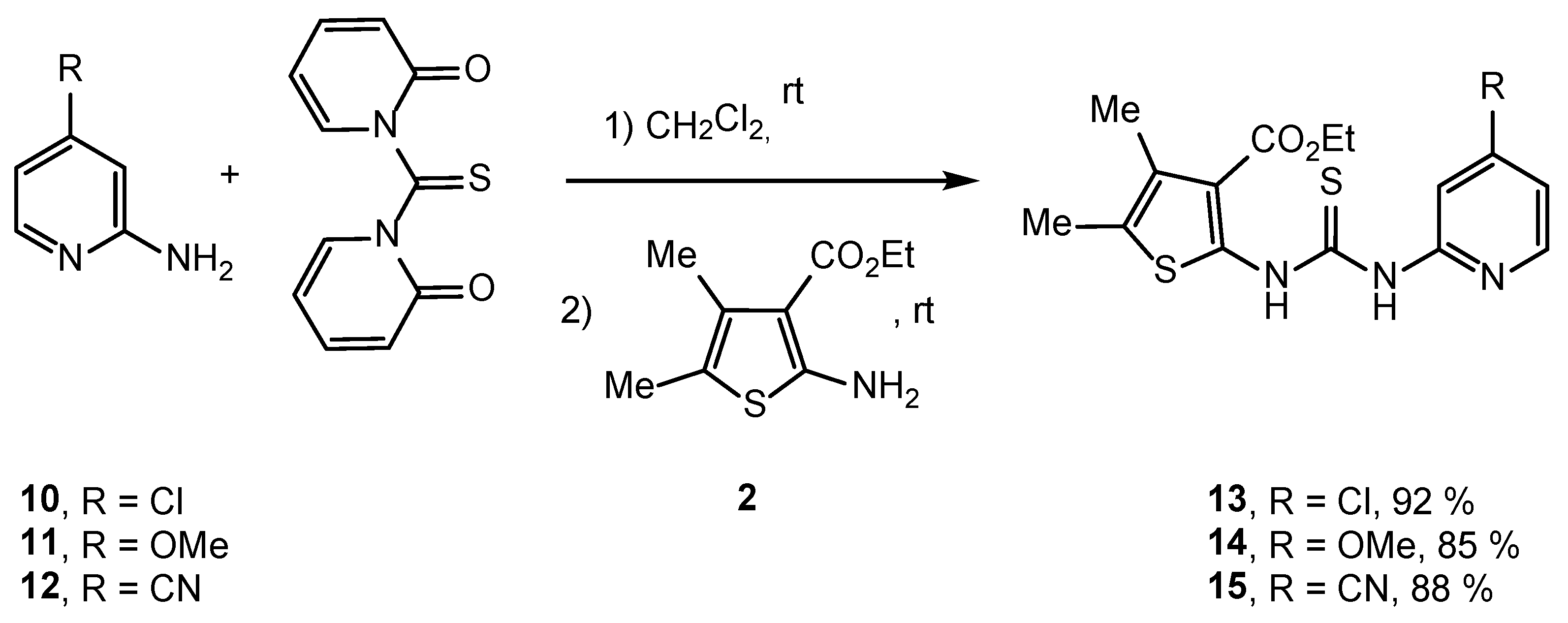
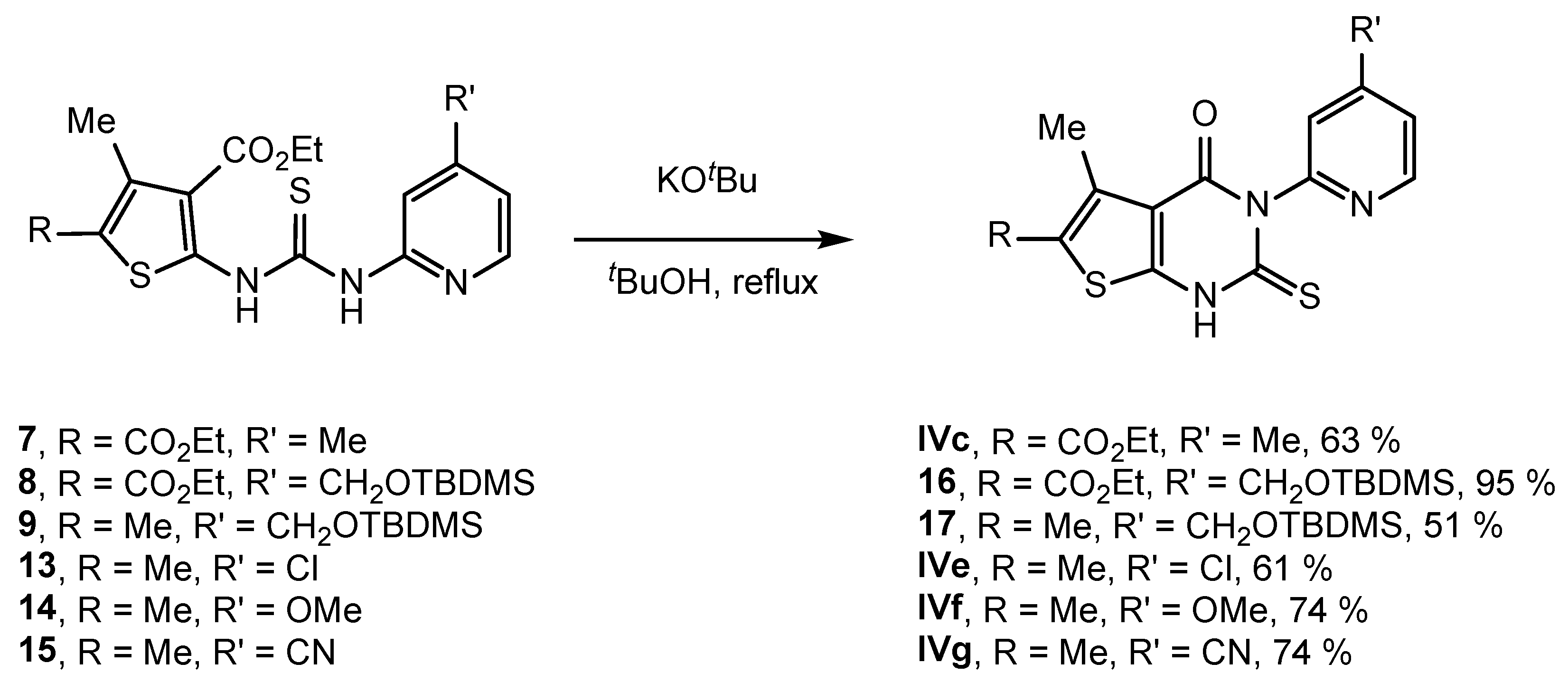
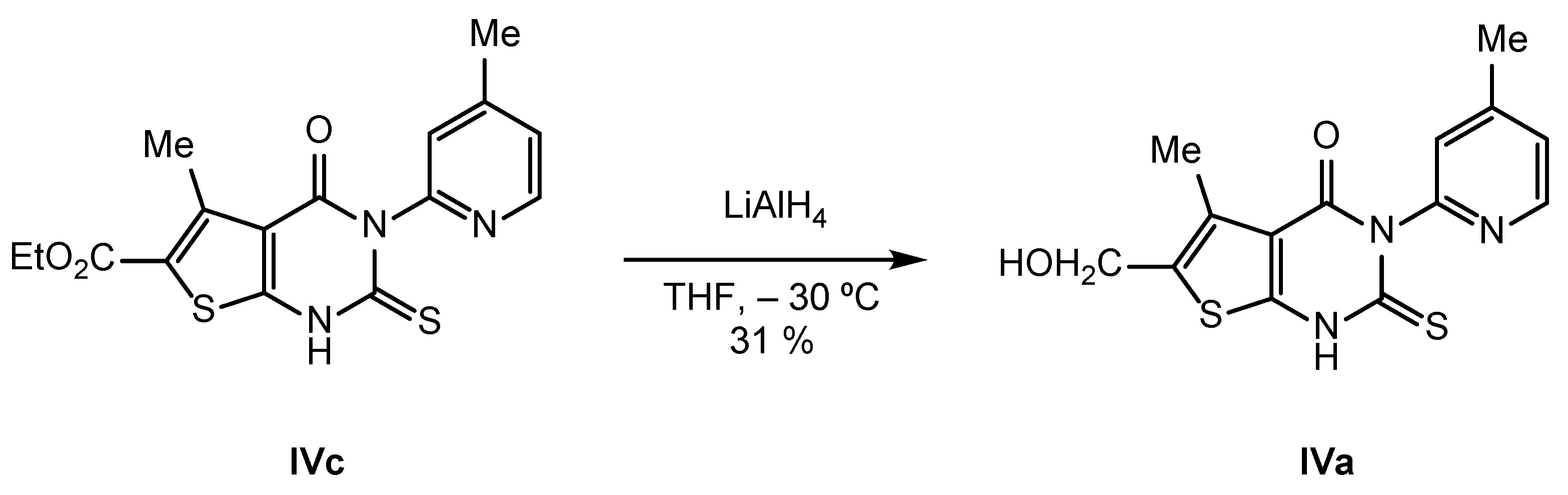
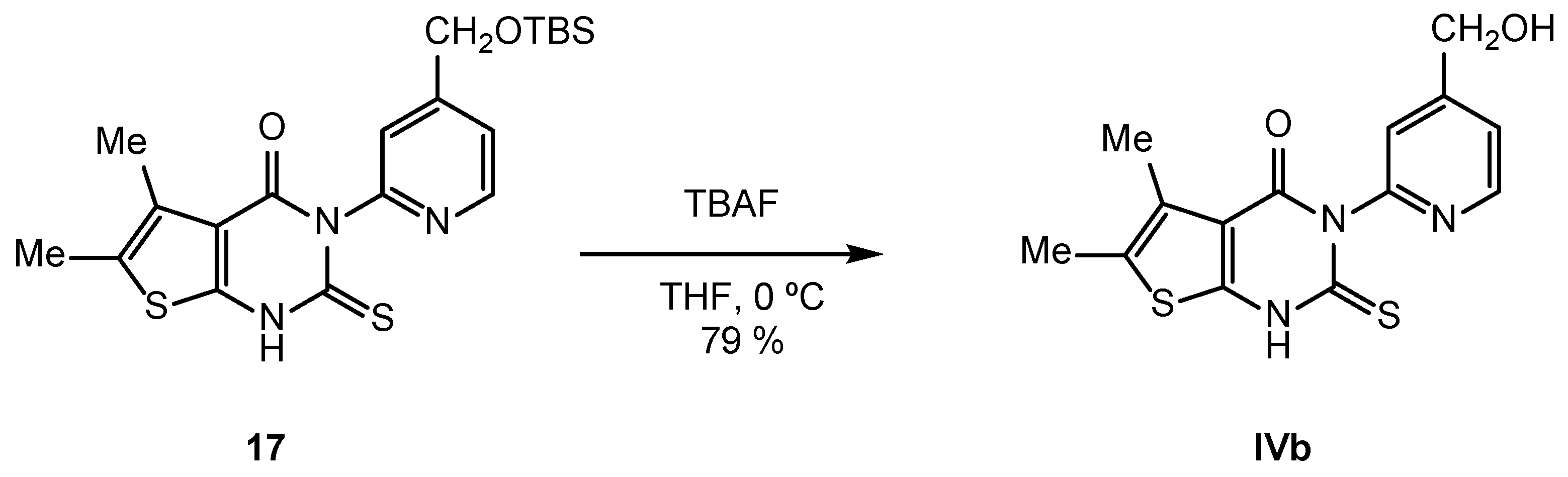
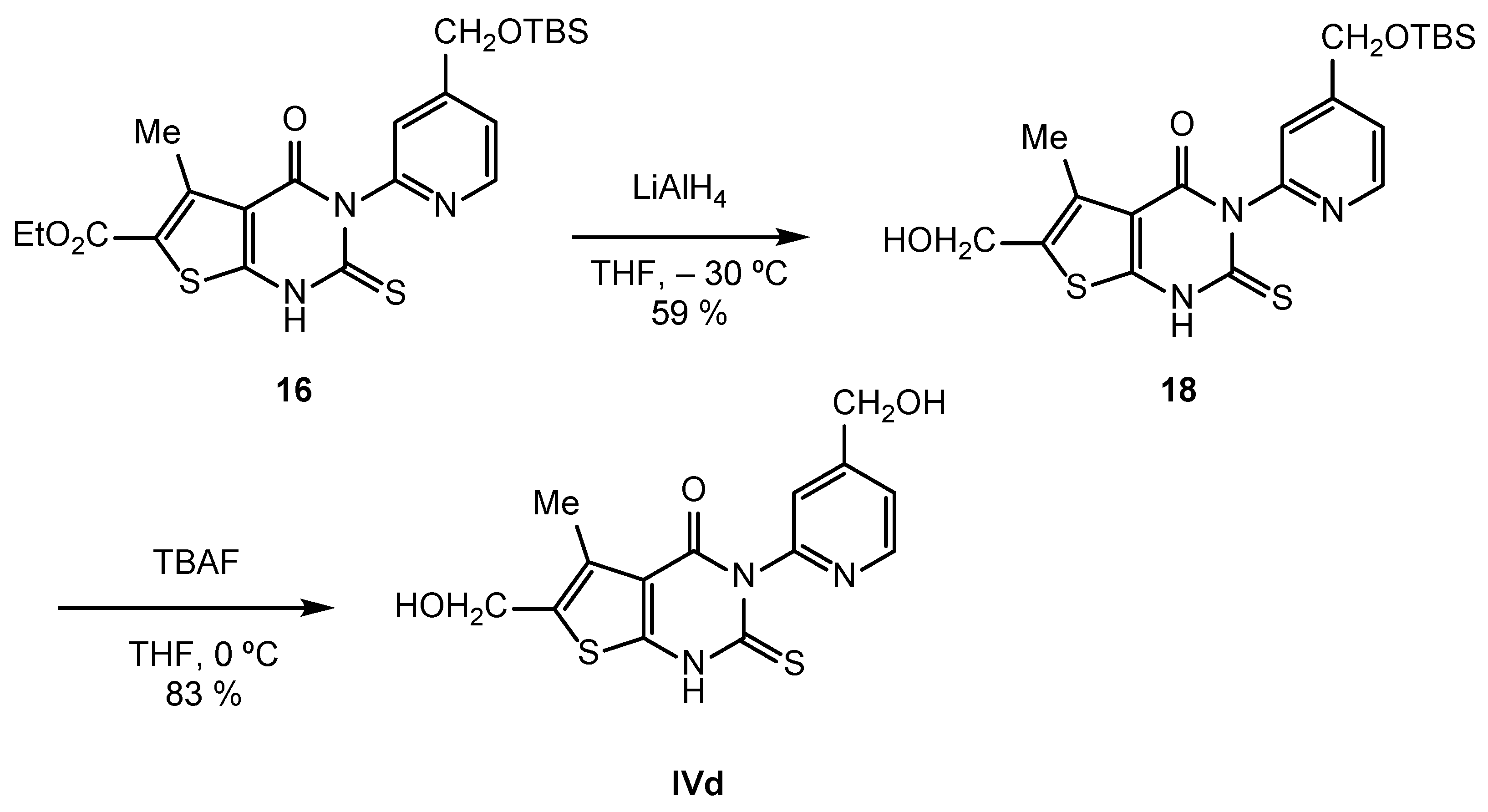
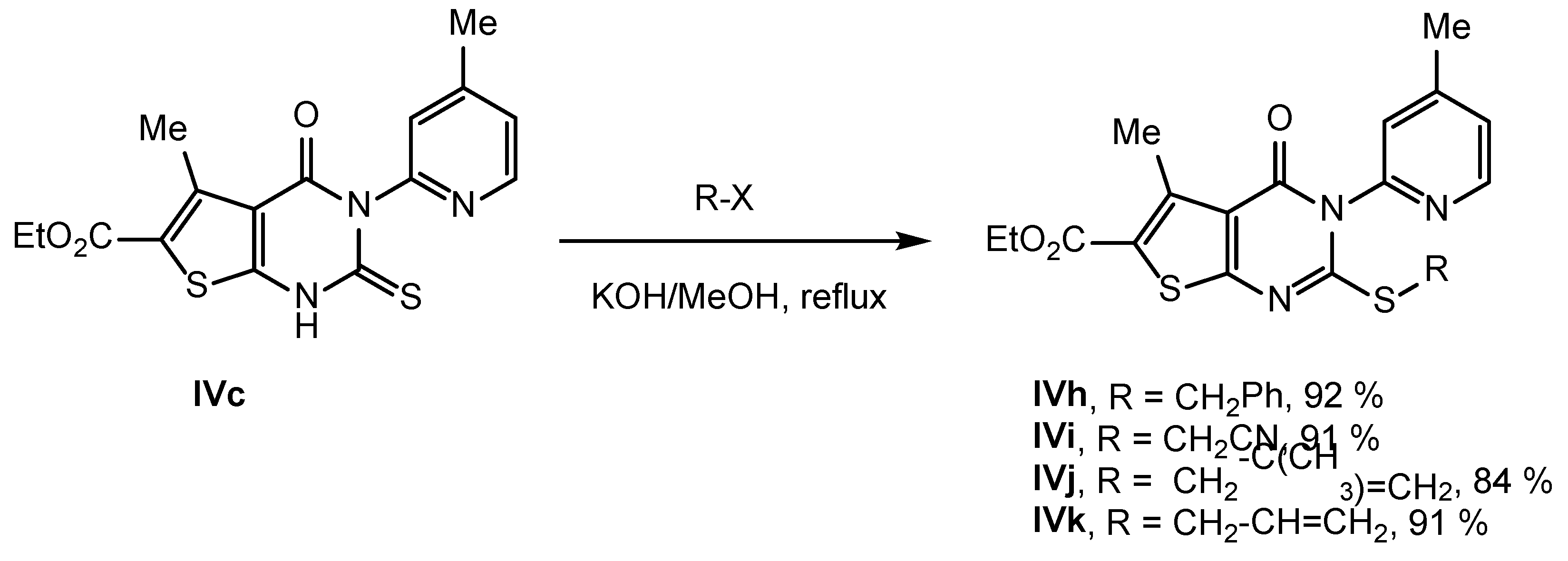
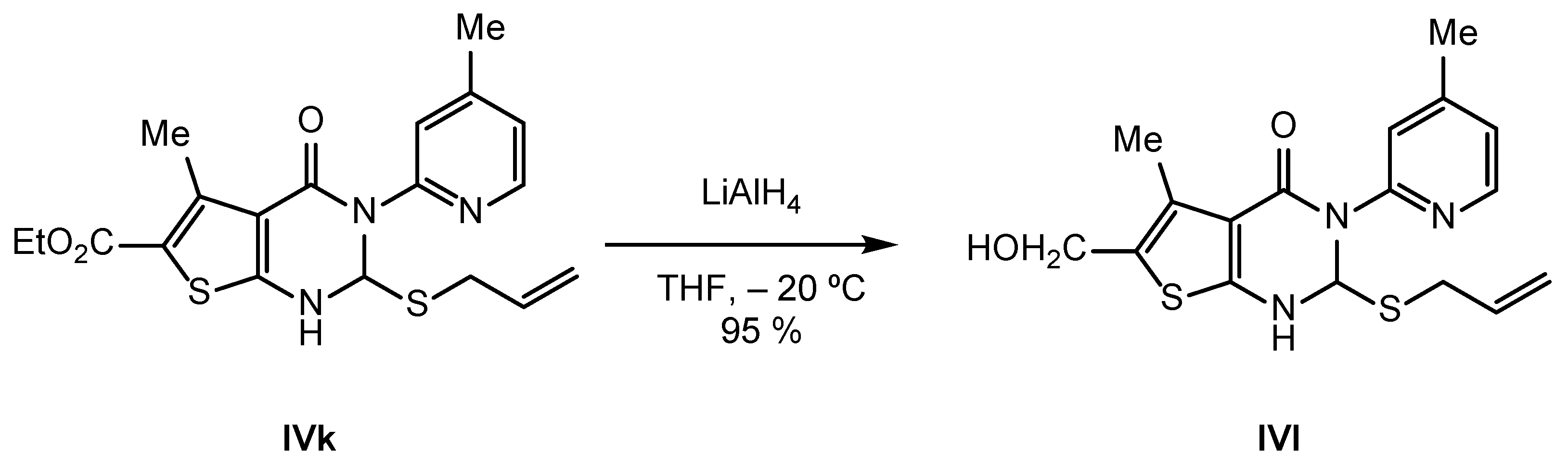
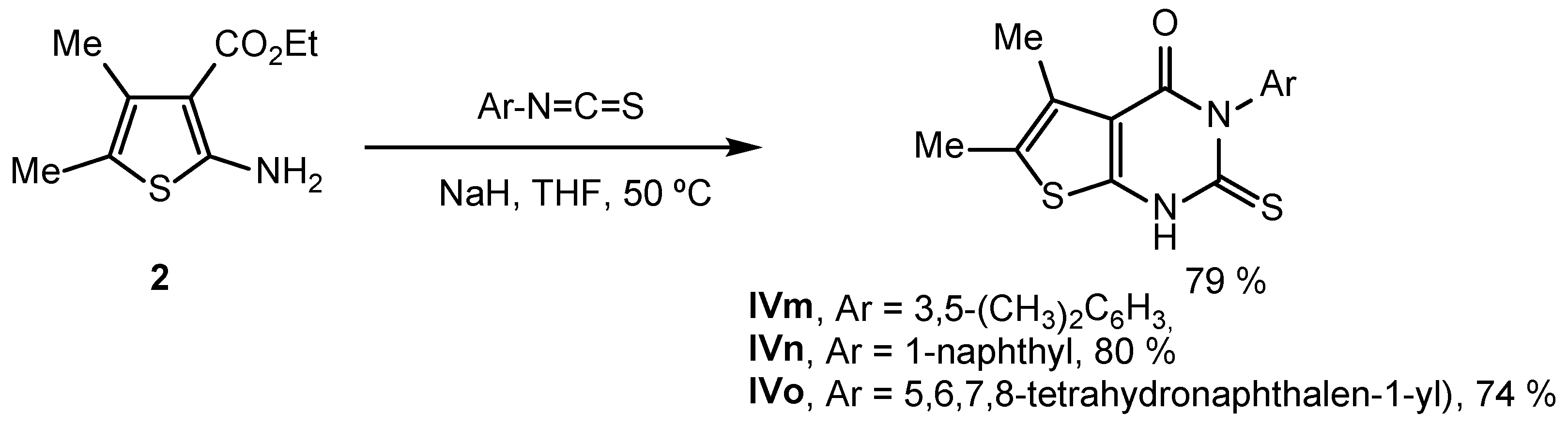
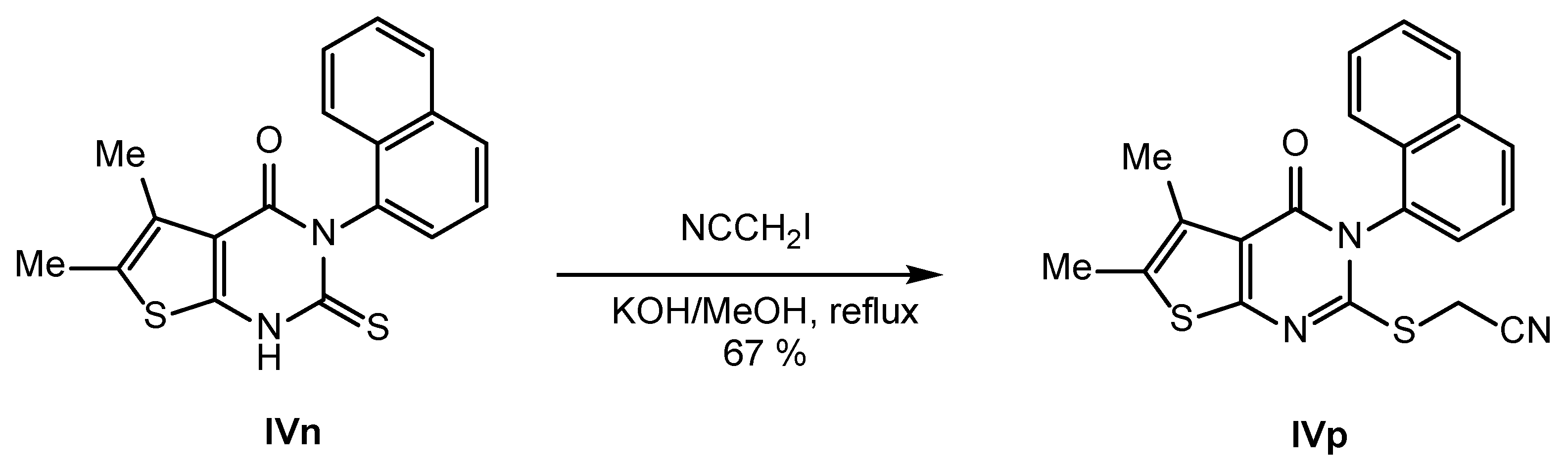
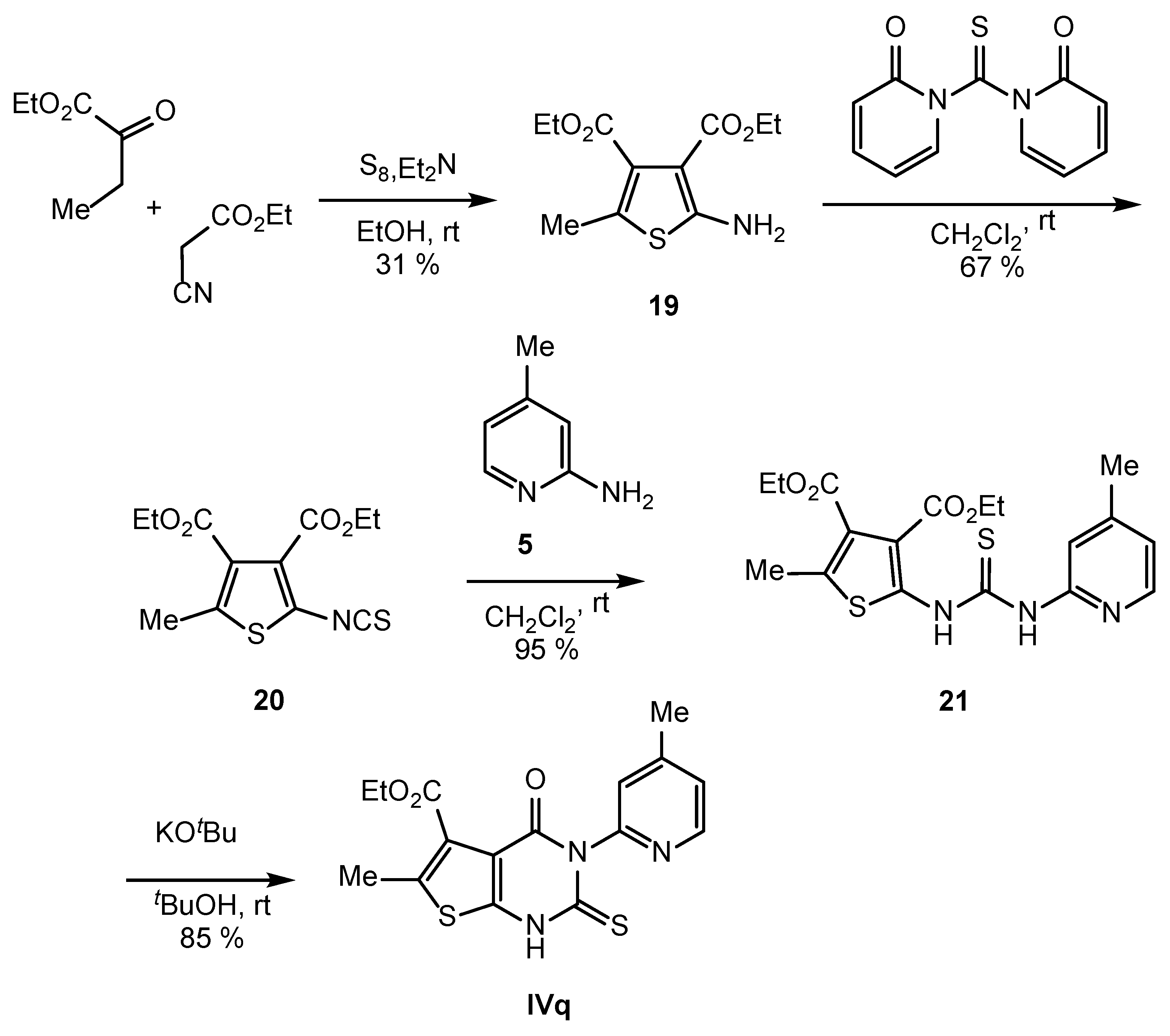
2.2. Synthesis of IVPC Analogues IVa–IVq
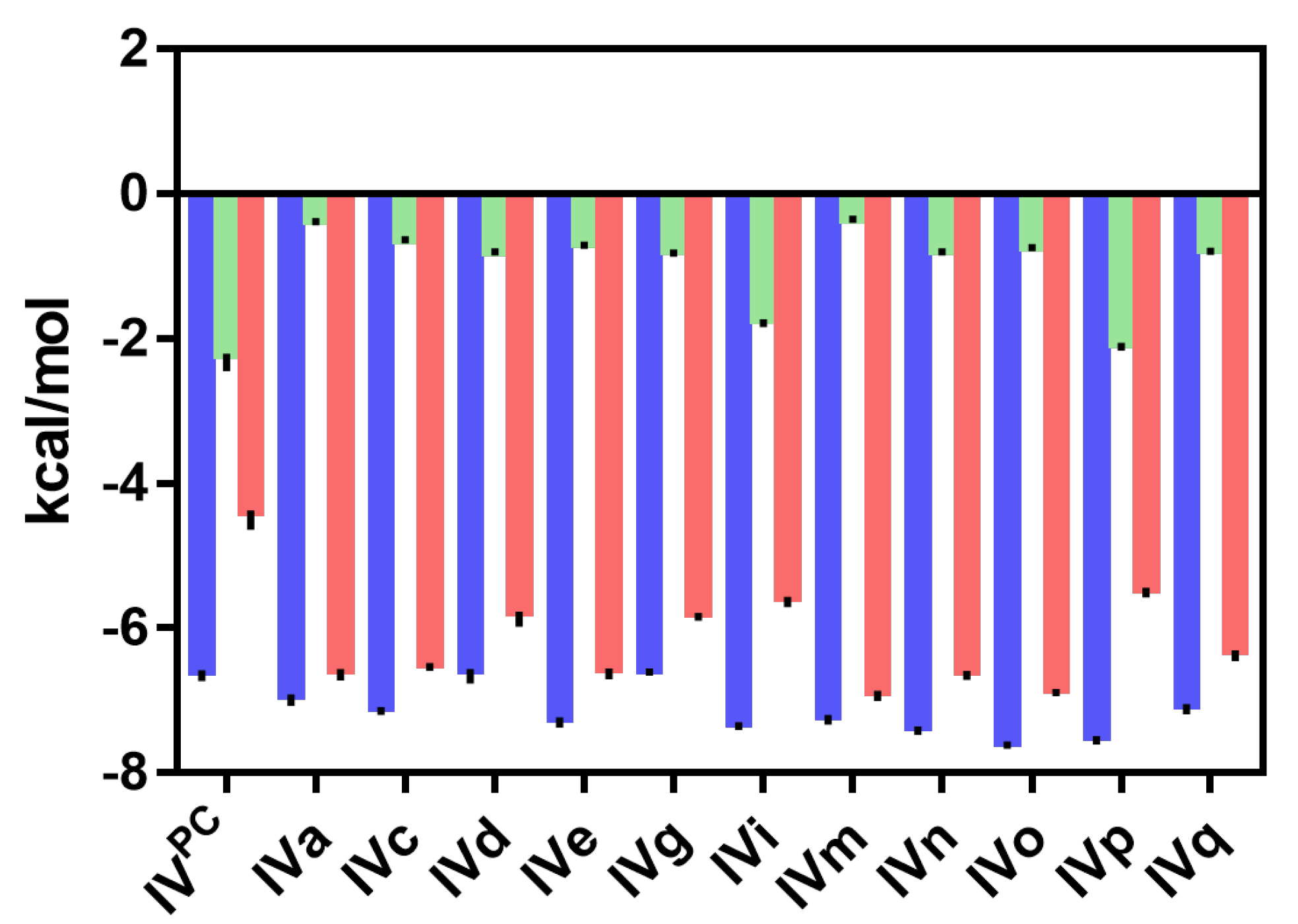
2.3. In Silico Calculation of the Affinity of the PAH-IVPC and PAH-IVx Complexes
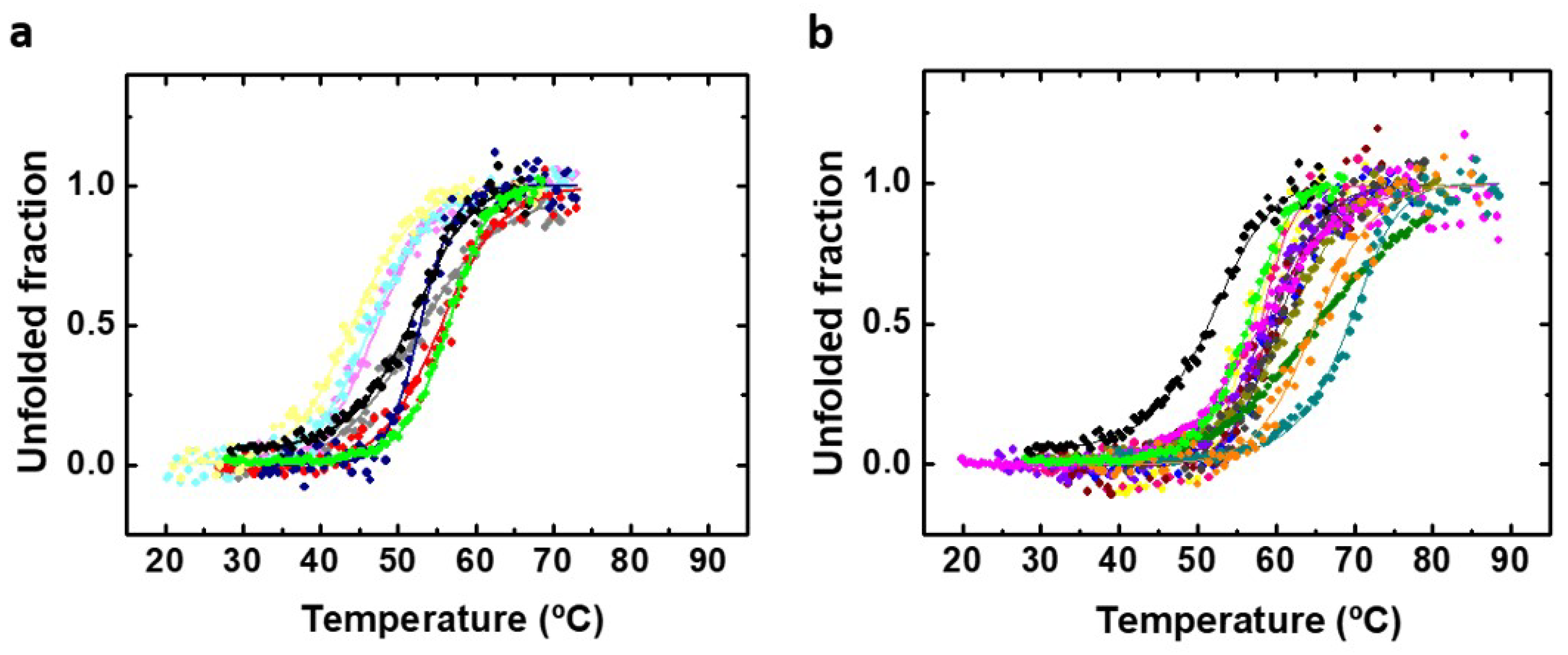
2.4. Actual Affinity of the PAH-IVx Complexes and its Effect on PAH Thermostability
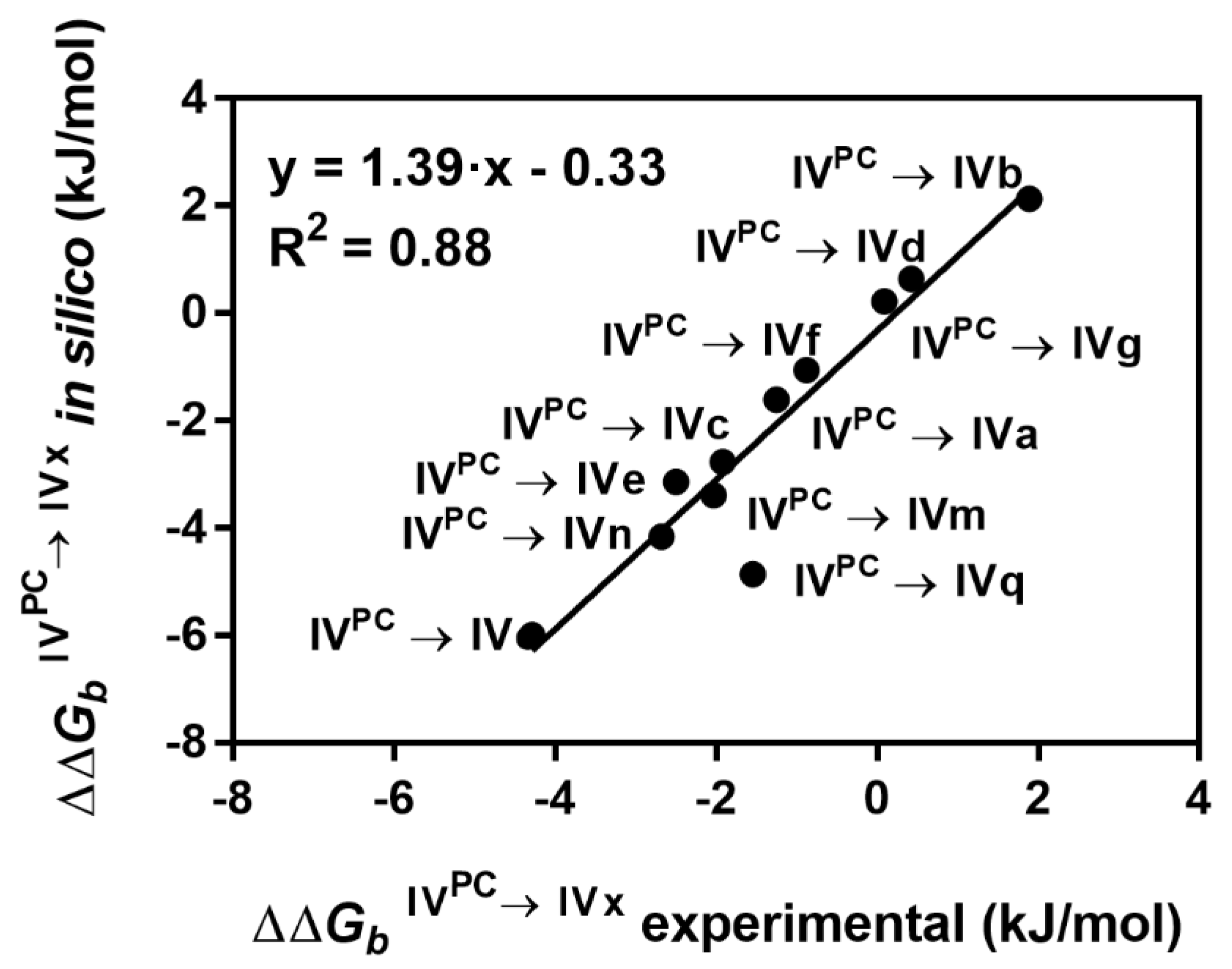
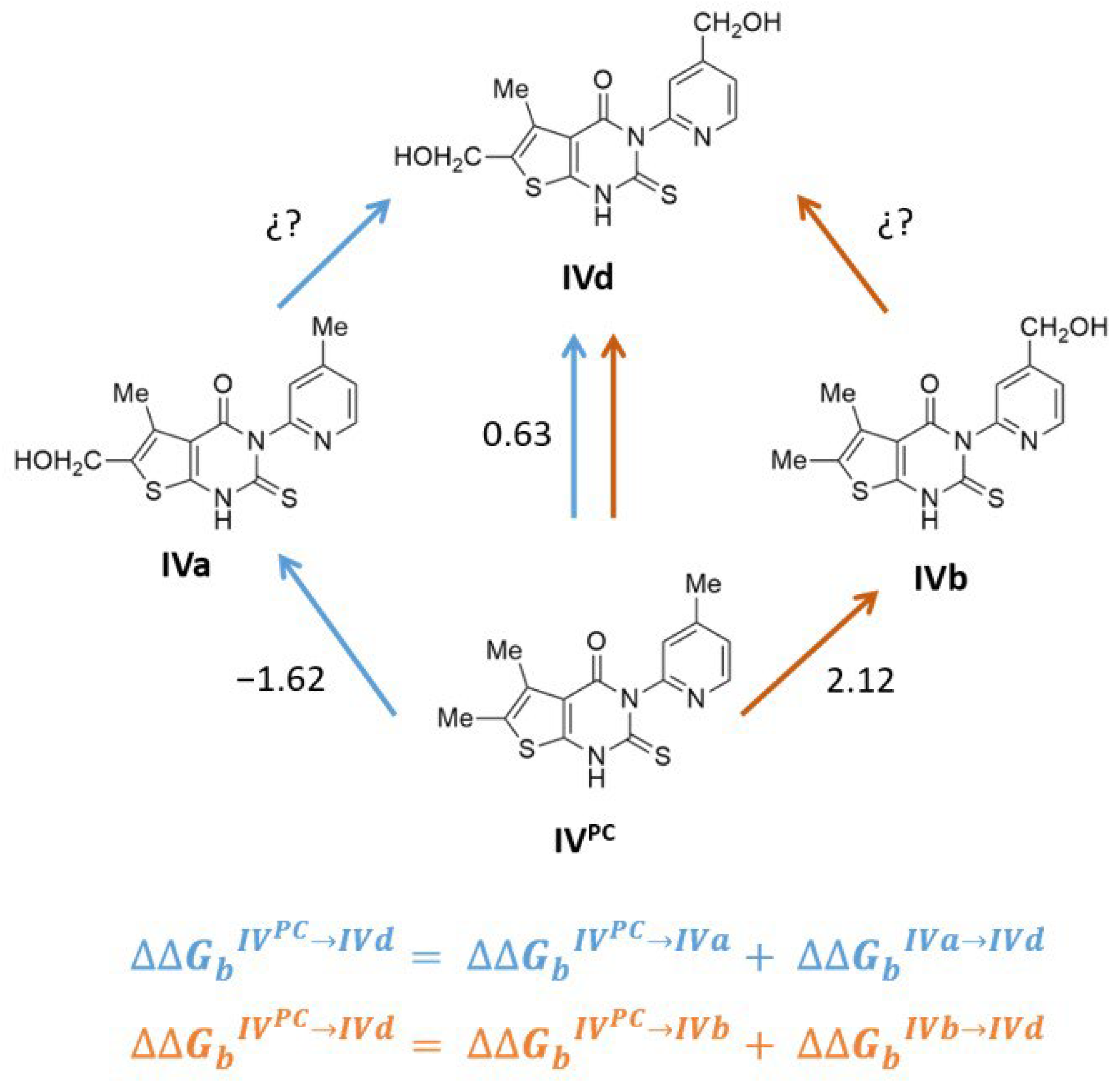
2.5. Usefulness of AFEC for the Rational Design of Better PAH Binders
3. Materials and Methods
3.1. Reagents and Chemicals
3.2. Parameterization of the Metal Center and IVPC Analogues, and Molecular Dynamics (MD) Preparation Setup for the Alchemical (AFEC) Simulations
3.3. Alchemical Free Energy Calculation (AFEC)
3.4. Expression and Purification of Human Recombinant PAH
3.5. Fluorescence Thermal Denaturation Measurements
3.6. Isothermal Titration Calorimetry (ITC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Scriver, C.R.; Kaufman, S. Hyperphenylalaninaemia: Phenylalanine hydroxylase deficiency. In The Metabolicand Molecular Basesof Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Slyetal, W.S., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 1667–1724. [Google Scholar]
- Güttler, F. Hyperphenylalaninemia: Diagnosis and classification of the various types of phenylalanine hydroxylase deficiency in childhood. Acta Paediatr Scand. 1980, 280, 180. [Google Scholar]
- Van Wegberg, A.M.J.; Macdonald, A.; Ahring, K.; BéLanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Bickel, H.; Gerrard, J.; Hickmans, E. Preliminary Communication. Lancet 1953, 262, 812–813. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.O. New era in treatment for phenylketonuria: Pharmacologic therapy with sapropterin dihydrochloride. Biol. Targets Ther. 2010, 4, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Pey, A.L.; Pérez, B.; Desviat, L.R.; Martínez, M.A.; Aguado, C.; Erlandsen, H.; Gámez, A.; Stevens, R.; Thórólfsson, M.; Ugarte, M.; et al. Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. Hum. Mutat. 2004, 24, 388–399. [Google Scholar] [CrossRef]
- Ho, G.; Christodoulou, J. Phenylketonuria: Translating research into novel therapies. Transl. Pediatr. 2014, 3, 49–62. [Google Scholar]
- Thomas, J.; Levy, H.; Amato, S.; Vockley, J.; Zori, R.; Dimmock, D.; Harding, C.O.; Bilder, D.A.; Weng, H.H.; Olbertz, J.; et al. Pegvaliase for the treatment of phenylketonuria: Results of a long-term phase 3 clinical trial program (PRISM). Mol. Genet. Metab. 2018, 124, 27–38. [Google Scholar] [CrossRef]
- Longo, N.; Dimmock, D.; Levy, H.; Viau, K.; Bausell, H.; Bilder, D.A.; Burton, B.; Gross, C.; Northrup, H.; Rohr, F.; et al. Evidence- and consensus-based recommendations for the use of pegvaliase in adults with phenylketonuria. Genet. Med. 2019, 21, 1851–1867. [Google Scholar] [CrossRef] [Green Version]
- Pey, A.L.; Ying, M.; Cremades, N.; Velázquez-Campoy, A.; Scherer, T.; Thöny, B.; Sancho, J.; Martínez, A. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J. Clin. Investig. 2008, 118, 2858–2867. [Google Scholar] [CrossRef] [Green Version]
- Torreblanca, R.; Lira-Navarrete, E.; Sancho, J.; Hurtado-Guerrero, R. Structural and Mechanistic Basis of the Interaction between a Pharmacological Chaperone and Human Phenylalanine Hydroxylase. ChemBioChem 2012, 13, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- De Ruiter, A.; Oostenbrink, C. Free energy calculations of protein–ligand interactions. Curr. Opin. Chem. Biol. 2011, 15, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Mobley, D.L.; Shirts, M.R.; Dixon, R.W.; Branson, K.; Pande, V.S. Alchemical free energy methods for drug discovery: Progress and challenges. Curr. Opin. Struct. Biol. 2011, 21, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, C.E.M.; Baumann, H.; Blum, A.; Böse, D.; Buchstaller, H.-P.; Burgdorf, L.; Cappel, D.; Chekler, E.; Czodrowski, P.; Dorsch, D.; et al. Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. J. Chem. Inf. Model. 2020, 60, 5457–5474. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Kim, B.R.; Lee, H.-G.; Kang, S.-B.; Sung, G.H.; Kim, J.-J.; Park, J.K.; Lee, S.-G.; Yoon, Y.-J. tert-Butoxide-Assisted Amidationof Estersunder Green Conditions. Synthesis 2012, 44, 42–50. [Google Scholar]
- Kim, S.; Yi, K.Y. 1,1′-Thiocarbonyldi-2,2′-pyridone. A new useful reagent for functional group conversions under essentially neutral conditions. J. Org. Chem. 1986, 51, 2613–2615. [Google Scholar] [CrossRef]
- Arya, D.P.; Bruice, T.C. Positively charged deoxynucleic methylthioureas: Synthesis and binding properties of pentameric thymidyl methylthiourea. J. Am. Chem. Soc. 1998, 120, 12419–12427. [Google Scholar] [CrossRef]
- Wang, K.; Li, J.; Degterev, A.; Hsu, E.; Yuan, J.; Yuan, C. Structure–activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5. Bioorganic Med. Chem. Lett. 2007, 17, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Gewald, K.; Schinke, E.; Böttcher, H. Heterocyclen aus CH-aciden Nitrilen, VIII. 2-Amino-thiophene aus methylenaktiven Nitrilen, Carbonylverbindungen und Schwefel. Eur. J. Inorg. Chem. 1966, 99, 94–100. [Google Scholar] [CrossRef]
- Briel, D.; Rybak, A.; Kronbach, C.; Unverferth, K. Substituted 2-Aminothiopen-derivatives: A potential new class of GluR6-Antagonists. Eur. J. Med. Chem. 2010, 45, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Conde-Giménez, M.; Sancho, J. Unravelling the Complex Denaturant and Thermal-Induced Unfolding Equilibria of Human Phenylalanine Hydroxylase. Int. J. Mol. Sci. 2021, 22, 6539. [Google Scholar] [CrossRef]
- Celej, M.S.; Montich, G.G.; Fidelio, G.D. Protein stability induced by ligand binding correlates with changes in protein flexibility. Protein Sci. 2003, 12, 1496–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claveria-Gimeno, R.; Vega, S.; Abian, O.; Velazquez-Campoy, A. A Look at Ligand Binding Thermodynamics in Drug Discovery. Expert Opin. Drug Discov. 2017, 12, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef]
- Tran, M.L.; Génisson, Y.; Ballereau, S.; Dehoux, C. Second-Generation Pharmacological Chaperones: Beyond Inhibitors. Molecules 2020, 25, 3145. [Google Scholar] [CrossRef] [PubMed]
- Hole, M.; Jorge-Finnigan, A.; Underhaug, J.; Teigen, K.; Martinez, A. Pharmacological Chaperones that Protect Tetrahydrobiopterin Dependent Aromatic Amino Acid Hydroxylases Through Different Mechanisms. Curr. Drug Targets 2016, 17, 1515–1526. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, ClinicalTranslation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S. AMBER2018; University of California: Los Angeles, CA, USA, 2018. [Google Scholar]
- Li, P.; Merz, J.K.M. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian09 (RevisionA02); Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Parsons, J.; Holmes, J.B.; Rojas, J.M.; Tsai, J.; Strauss, C.E.M. Practical conversion from torsion space to Cartesian space forin silico protein synthesis. J. Comput. Chem. 2005, 26, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Besler, B.H.; Merz, K.M., Jr.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Woods, R.; Chappelle, R. Restrained electrostatic potential atomic partial charges for condensed-phase simulations of carbohydrates. J. Mol. Struct. THEOCHEM 2000, 527, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Strain fluctuations and elastic constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Fukunishi, H.; Watanabe, O.; Takada, S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: Application to protein structure prediction. J. Chem. Phys. 2002, 116, 9058–9067. [Google Scholar] [CrossRef]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Voth, G.A. Smart resolution replica exchange: An efficient algorithm for exploring complex energy landscapes. J. Chem. Phys. 2007, 126, 045106. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Dashti, D.S.; Roitberg, A.E. Computing alchemical free energy differences with Hamiltonian replica exchange molecular dynamics (H-REMD) simulations. J. Chem. Theory Comput. 2011, 79, 2721–2727. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Shirts, M.R.; Chodera, J.D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; Volume XVIII. [Google Scholar]
- Sancho, J. The stability of 2-state, 3-state and more-state proteins from simple spectroscopic techniques… plus the structure of the equilibrium intermediates at the same time. Arch. Biochem. Biophys. 2013, 531, 4–13. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Ohtaka, H.; Nezami, A.; Muzammil, S.; Freire, E. Isothermal Titration Calorimetry. Curr. Protoc. Cell Biol. 2004, 23, 17.8.1–17.8.24. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | MW (g/mol) | Modified Heterocycle |
|---|---|---|---|
| IVPC |  | 303.4 | - |
| IVa |  | 319.4 | Thiophene |
| IVb |  | 319.4 | Pyridine |
| IVc |  | 361.4 | Thiophene |
| IVd |  | 335.4 | Thiophene and Pyridine |
| IVe |  | 323.8 | Pyridine |
| IVf |  | 319.4 | Pyridine |
| IVg |  | 314.4 | Pyridine |
| IVh |  | 451.6 | Thiophene and Pyrimidine |
| IVi |  | 400.5 | Thiophene and Pyrimidine |
| IVj |  | 415.5 | Thiophene and Pyrimidine |
| IVk |  | 401.5 | Thiophene and Pyrimidine |
| IVl |  | 359.5 | Thiophene and Pyrimidine |
| IVm |  | 316.4 | Pyridine (substitution) |
| IVn |  | 338.5 | Pyridine (substitution) |
| IVo |  | 342.5 | Pyridine (substitution) |
| IVp |  | 379.5 | Pyridine (substitution) and Pyrimidine |
| IVq |  | 361.4 | Thiophene |
| Compound | In Silico ∆∆Gb IVPC→IVx (kJ/mol) a | Experimental ∆∆Gb (kJ/mol) b | ΔTm (°C) c | |
|---|---|---|---|---|
| with FeII | with FeIII | |||
| IVPC | - | - | - | 5.0 ± 0.6 |
| IVa | −1.62 ± 0.18 | −0.66 ± 0.25 | −1.25 ± 1.19 | 5.5 ± 0.8 |
| IVb | 2.12 ± 0.79 | −0.08 ± 0.23 | 1.89 ± 1.60 | 1.2 ± 0.9 |
| IVc | −2.78 ± 1.70 | −1.65 ± 0.93 | −1.92 ± 1.31 | 6.5 ± 0.8 |
| IVd | 0.63 ± 0.37 | −0.73 ± 0.19 | 0.42 ± 1.29 | 8.6 ± 0.5 |
| IVe | −3.14 ± 0.19 | −3.98 ± 0.09 | −2.50 ± 1.12 | 8.0 ± 1.4 |
| IVf | −1.06 ± 0.37 | −3.04 ± 0.33 | −0.88 ± 1.25 | 4.1 ± 0.8 |
| IVg | 0.21 ± 0.37 | −4.01 ± 0.37 | 0.09 ± 1.17 | 7.3 ± 0.8 |
| IVh | n.d. c | n.d.c | −0.10 ± 1.13 | −4.1 ± 0.7 |
| IVi | 0.64 ± 3.21 | −22.94 ± 4.36 | −2.46 ± 1.55 | 14.1 ± 0.6 |
| IVj | n.d. c | −9.52 ± 8.95 | 1.65 ± 1.10 | −4.9 ± 0.8 |
| IVk | 3.01 ± 0.62 | n.d. c | −0.43 ± 1.14 | −7.4 ± 0.6 |
| IVl | 8.36 ± 2.64 | n.d. c | 0.50 ± 1.08 | 1.3 ± 0.6 |
| IVm | −3.39 ± 0.80 | −3.28 ± 0.93 | −2.03 ± 1.26 | 10.0 ± 0.9 |
| IVn | −4.17 ± 0.89 | −7.33 ± 0.32 | −2.68 ± 1.27 | 13.6 ± 1.4 |
| IVo | −5.99 ± 0.64 | −3.17 ± 1.09 | −4.28 ± 1.37 | 8.8 ± 0.6 |
| IVp | −1.00 ± 2.87 | −11.85 ± 3.02 | −3.57 ± 1.10 | 17.9 ± 0.9 |
| IVq | −4.86 ± 0.76 | −3.86 ± 0.31 | −1.54 ± 1.32 | 6.7 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conde-Giménez, M.; Galano-Frutos, J.J.; Galiana-Cameo, M.; Mahía, A.; Victor, B.L.; Salillas, S.; Velázquez-Campoy, A.; Brito, R.M.M.; Gálvez, J.A.; Díaz-de-Villegas, M.D.; et al. Alchemical Design of Pharmacological Chaperones with Higher Affinity for Phenylalanine Hydroxylase. Int. J. Mol. Sci. 2022, 23, 4502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094502
Conde-Giménez M, Galano-Frutos JJ, Galiana-Cameo M, Mahía A, Victor BL, Salillas S, Velázquez-Campoy A, Brito RMM, Gálvez JA, Díaz-de-Villegas MD, et al. Alchemical Design of Pharmacological Chaperones with Higher Affinity for Phenylalanine Hydroxylase. International Journal of Molecular Sciences. 2022; 23(9):4502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094502
Chicago/Turabian StyleConde-Giménez, María, Juan José Galano-Frutos, María Galiana-Cameo, Alejandro Mahía, Bruno L. Victor, Sandra Salillas, Adrián Velázquez-Campoy, Rui M. M. Brito, José Antonio Gálvez, María D. Díaz-de-Villegas, and et al. 2022. "Alchemical Design of Pharmacological Chaperones with Higher Affinity for Phenylalanine Hydroxylase" International Journal of Molecular Sciences 23, no. 9: 4502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094502