
A Bioengineered In Vitro Model to Assess AAV-Based Gene Therapies for Cyclic GMP-Related Disorders



Abstract
:1. Introduction
2. Results
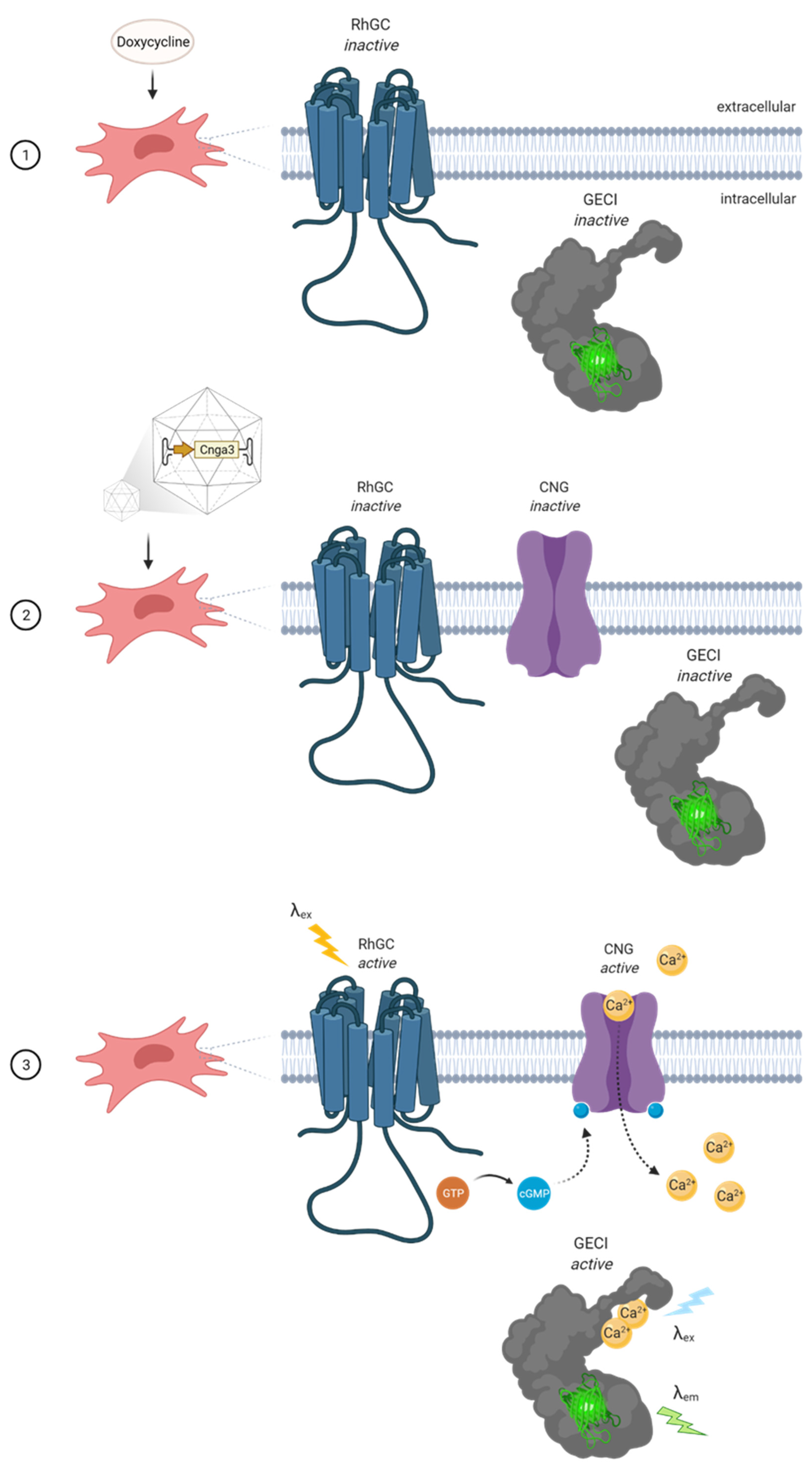
2.1. The Concept of the Cell-Based In Vitro Potency System
2.2. Validation of Basic Functions
2.3. Testing AAV Vectors with CNGA3 Cargo
2.4. Combined AAV Vector Testing with CNGA3 and PDE6A Cargo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of New Hela Cell Lines
4.3. Gene Expression Analysis
4.4. RhGC Illumination and GECI Recordings
4.5. Immunocytochemistry and Imaging
4.6. AAV Vector Production and Cell Transduction
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michalakis, S.; Gerhardt, M.; Rudolph, G.; Priglinger, S.; Priglinger, C. Achromatopsia: Genetics and Gene Therapy. Mol. Diagn. Ther. 2022, 26, 51. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlfriedel, R.; Tanimoto, N.; Schön, C.; Sothilingam, V.; Garrido, M.G.; Beck, S.C.; Huber, G.; Biel, M.; Seeliger, M.W.; Michalakis, S. AAV-mediated gene supplementation therapy in achromatopsia type 2: Preclinical data on therapeutic time window and long-term effects. Front. Neurosci. 2017, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Mühlfriedel, R.; Tanimoto, N.; Krishnamoorthy, V.; Koch, S.; Fischer, M.D.; Becirovic, E.; Bai, L.; Huber, G.; Beck, S.C.; et al. Restoration of cone vision in the CNGA3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 2010, 18, 2057–2063. [Google Scholar] [CrossRef]
- Banin, E.; Gootwine, E.; Obolensky, A.; Ezra-Elia, R.; Ejzenberg, A.; Zelinger, L.; Honig, H.; Rosov, A.; Yamin, E.; Sharon, D.; et al. Gene Augmentation Therapy Restores Retinal Function and Visual Behavior in a Sheep Model of CNGA3 Achromatopsia. Mol. Ther. 2015, 23, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Komáromy, A.M.; Rowlan, J.S.; Corr, A.T.P.; Reinstein, S.L.; Boye, S.L.; Cooper, A.E.; Gonzalez, A.; Levy, B.; Wen, R.; Hauswirth, W.W.; et al. Transient Photoreceptor Deconstruction by CNTF Enhances rAAV-Mediated Cone Functional Rescue in Late Stage CNGB3-Achromatopsia. Mol. Ther. 2013, 21, 1131. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, L.S.; Xu, J.; Pearson, R.A.; Smith, A.J.; Bainbridge, J.W.; Morris, L.M.; Fliesler, S.J.; Ding, X.Q.; Ali, R.R. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 2011, 20, 3161–3175. [Google Scholar] [CrossRef]
- Pang, J.J.; Deng, W.T.; Dai, X.; Lei, B.; Everhart, D.; Umino, Y.; Li, J.; Zhang, K.; Mao, S.; Boye, S.L.; et al. AAV-Mediated Cone Rescue in a Naturally Occurring Mouse Model of CNGA3-Achromatopsia. PLoS ONE 2012, 7, e35250. [Google Scholar] [CrossRef] [Green Version]
- Komáromy, A.M.; Alexander, J.J.; Rowlan, J.S.; Garcia, M.M.; Chiodo, V.A.; Kaya, A.; Tanaka, J.C.; Acland, G.M.; Hauswirth, W.W.; Aguirre, G.D. Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 2010, 19, 2581–2593. [Google Scholar] [CrossRef]
- Pavlou, M.; Schön, C.; Occelli, L.M.; Rossi, A.; Meumann, N.; Boyd, R.F.; Bartoe, J.T.; Siedlecki, J.; Gerhardt, M.J.; Babutzka, S.; et al. Novel AAV capsids for intravitreal gene therapy of photoreceptor disorders. EMBO Mol. Med. 2021, 13, e13392. [Google Scholar] [CrossRef]
- Keiser, N.W.; Yan, Z.; Zhang, Y.; Lei-Butters, D.C.M.; Engelhardt, J.F. Unique Characteristics of AAV1, 2, and 5 Viral Entry, Intracellular Trafficking, and Nuclear Import Define Transduction Efficiency in HeLa Cells. Hum. Gene Ther. 2011, 22, 1433–1444. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.H.; Coates, C.J.; George, A.L. PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. 2007, 15, 139–145. [Google Scholar] [CrossRef]
- Scheib, U.; Stehfest, K.; Gee, C.E.; Körschen, H.G.; Fudim, R.; Oertner, T.G.; Hegemann, P. The rhodopsin-guanylyl cyclase of the aquatic fungus Blastocladiella emersonii enables fast optical control of cGMP signaling. Sci. Signal. 2015, 8, rs8. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D.; et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 2016, 5, e12727. [Google Scholar] [CrossRef]
- Gossen, M.; Freundlieb, S.; Bender, G.; Müller, G.; Hillen, W.; Bujard, H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995, 268, 1766–1769. [Google Scholar] [CrossRef]
- Yao, F.; Svensjö, T.; Winkler, T.; Lu, M.; Eriksson, C.; Eriksson, E. Tetracycline repressor, tetR, rather than the tetR-mammalian cell transcription factor fusion derivatives, regulates inducible gene expression in mammalian cells. Hum. Gene Ther. 1998, 9, 1939–1950. [Google Scholar] [CrossRef]
- Cellular & Gene Therapy Guidances|FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/biologics-guidances/cellular-gene-therapy-guidances (accessed on 21 March 2022).
- Gerstner, A.; Zong, X.; Hofmann, F.; Biel, M. Molecular cloning and functional characterization of a new modulatory cyclic nucleotide-gated channel subunit from mouse retina. J. Neurosci. 2000, 20, 1324–1332. [Google Scholar] [CrossRef]
- Wu, N.; Nishioka, W.K.; Derecki, N.C.; Maher, M.P. High-throughput-compatible assays using a genetically-encoded calcium indicator. Sci. Rep. 2019, 9, 12692. [Google Scholar] [CrossRef] [Green Version]
- Biel, M. Cyclic Nucleotide-regulated Cation Channels. J. Biol. Chem. 2009, 284, 9017. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, N.A.; Akopov, S.B.; Didych, D.A.; Nikolaev, L.G. In trans promoter activation by enhancers in transient transfection. Gene 2017, 603, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Goc, A.; Chami, M.; Lodowski, D.T.; Bosshart, P.; Moiseenkova-Bell, V.; Baehr, W.; Engel, A.; Palczewski, K. Structural characterization of the rod cGMP phosphodiesterase 6. J. Mol. Biol. 2010, 401, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An expanded palette of genetically encoded Ca2+ indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Rieser, R.; Koch, J.; Faccioli, G.; Richter, K.; Menzen, T.; Biel, M.; Winter, G.; Michalakis, S. Comparison of Different Liquid Chromatography-Based Purification Strategies for Adeno-Associated Virus Vectors. Pharmaceutics 2021, 13, 748. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward Primer | Reverse Primer | Annealing T °C |
|---|---|---|---|
| eGFP | CGACCACTACCAGCAGAACAC | TTCTCGTTGGGGTCTTTGCTCAG | 62 |
| ITR2 | GGAACCCCTAGTGATGGAGTT | CGGCCTCAGTGAGCGA | 58 |
| GCaMP6s | CCCGACAACCACTACCTGAG | GTCCATGCCGAGAGTGATCC | 60 |
| jRCaMP1a | AAGACAGGTCACGCAGTCAG | GAGTGTAACCACGCAGACCA | 60 |
| jRCaMP1b | GGAATAAGTGGGGTCACGCA | GAGTGTAACCACGCAGACCA | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlou, M.; Babutzka, S.; Michalakis, S. A Bioengineered In Vitro Model to Assess AAV-Based Gene Therapies for Cyclic GMP-Related Disorders. Int. J. Mol. Sci. 2022, 23, 4538. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094538
Pavlou M, Babutzka S, Michalakis S. A Bioengineered In Vitro Model to Assess AAV-Based Gene Therapies for Cyclic GMP-Related Disorders. International Journal of Molecular Sciences. 2022; 23(9):4538. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094538
Chicago/Turabian StylePavlou, Marina, Sabrina Babutzka, and Stylianos Michalakis. 2022. "A Bioengineered In Vitro Model to Assess AAV-Based Gene Therapies for Cyclic GMP-Related Disorders" International Journal of Molecular Sciences 23, no. 9: 4538. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094538