
Demystifying the Neuroprotective Role of Neuropeptides in Parkinson’s Disease: A Newfangled and Eloquent Therapeutic Perspective

 , , and
, , and 
Abstract
:1. Introduction
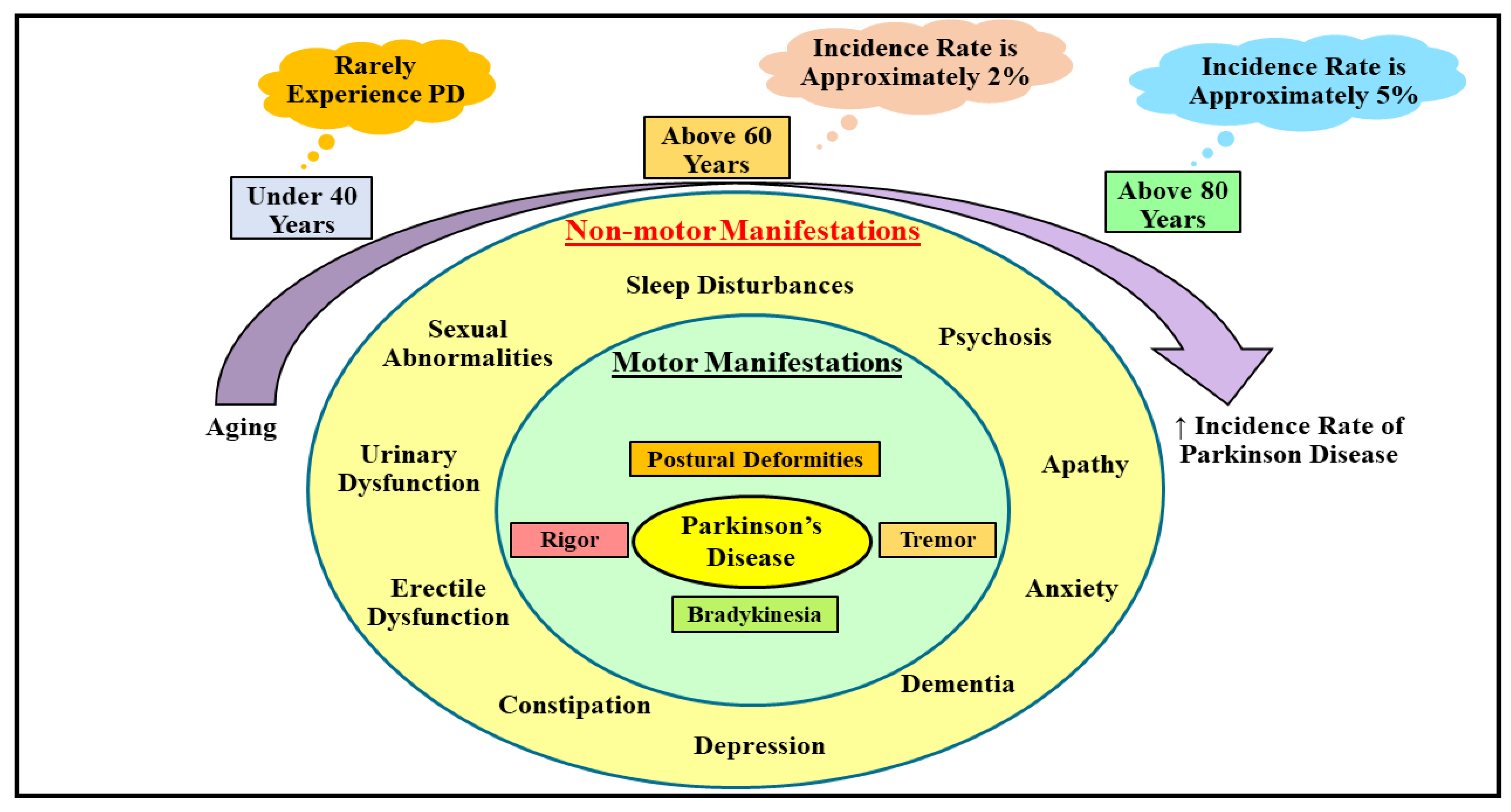
2. Understanding the Etiopathogenic Pathways Underlying Parkinson’s Disease
2.1. Understanding the Etiological Processes Underlying Parkinson’s Disease
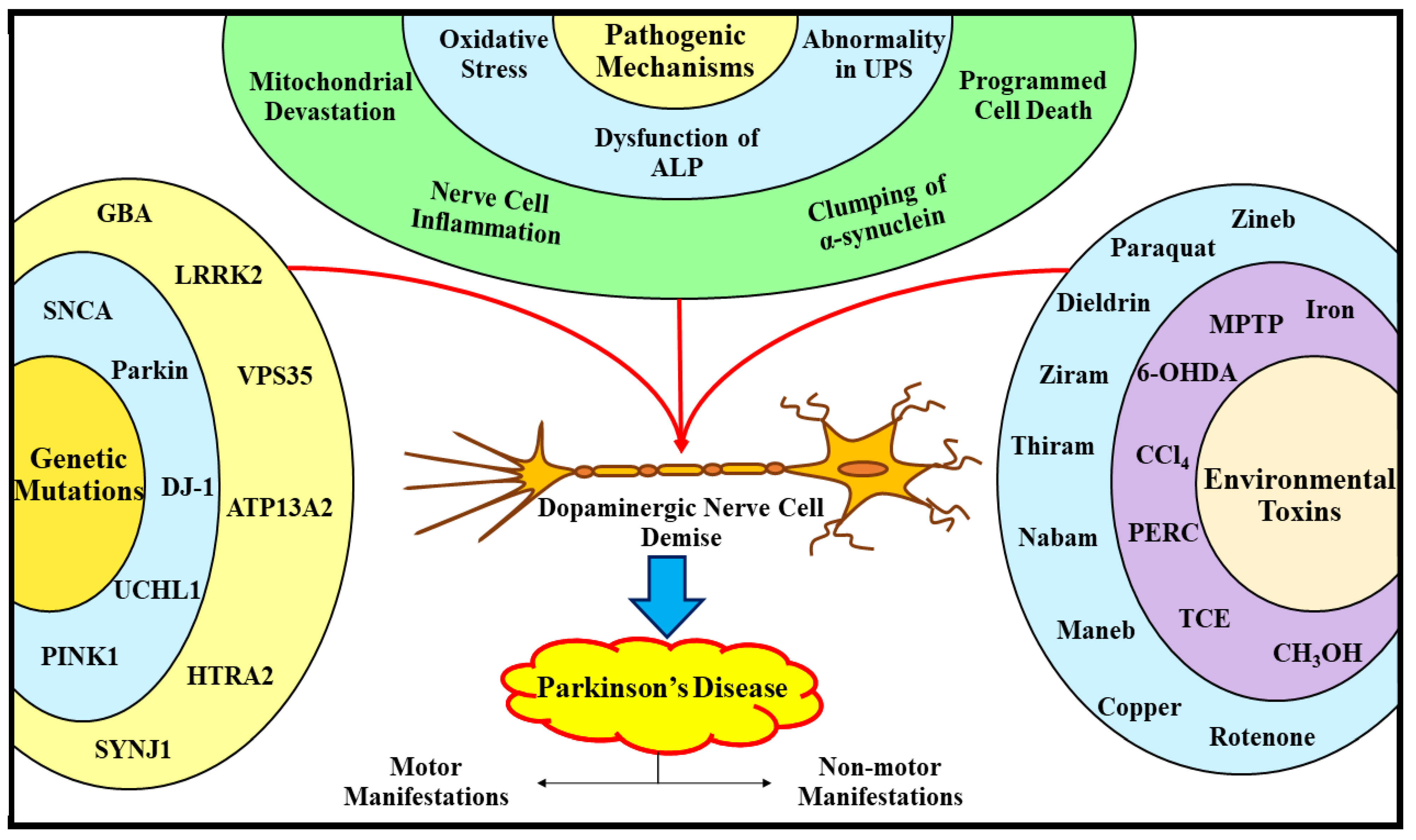
2.2. Understanding the Pathogenic Processes Underlying Parkinson’s Disease
2.2.1. Oxidative Stress and Parkinson’s Disease
2.2.2. Autophagy Lysosomal Pathway Dysfunction and Parkinson’s Disease
2.2.3. Ubiquitin–Proteasome System Dysfunction and Parkinson’s Disease
2.2.4. Mitochondrial Devastation and Parkinson’s Disease
2.2.5. Apoptosis, Nerve Cell Inflammation, and Parkinson’s Disease
2.2.6. α-Synuclein Clumping and Parkinson’s Disease
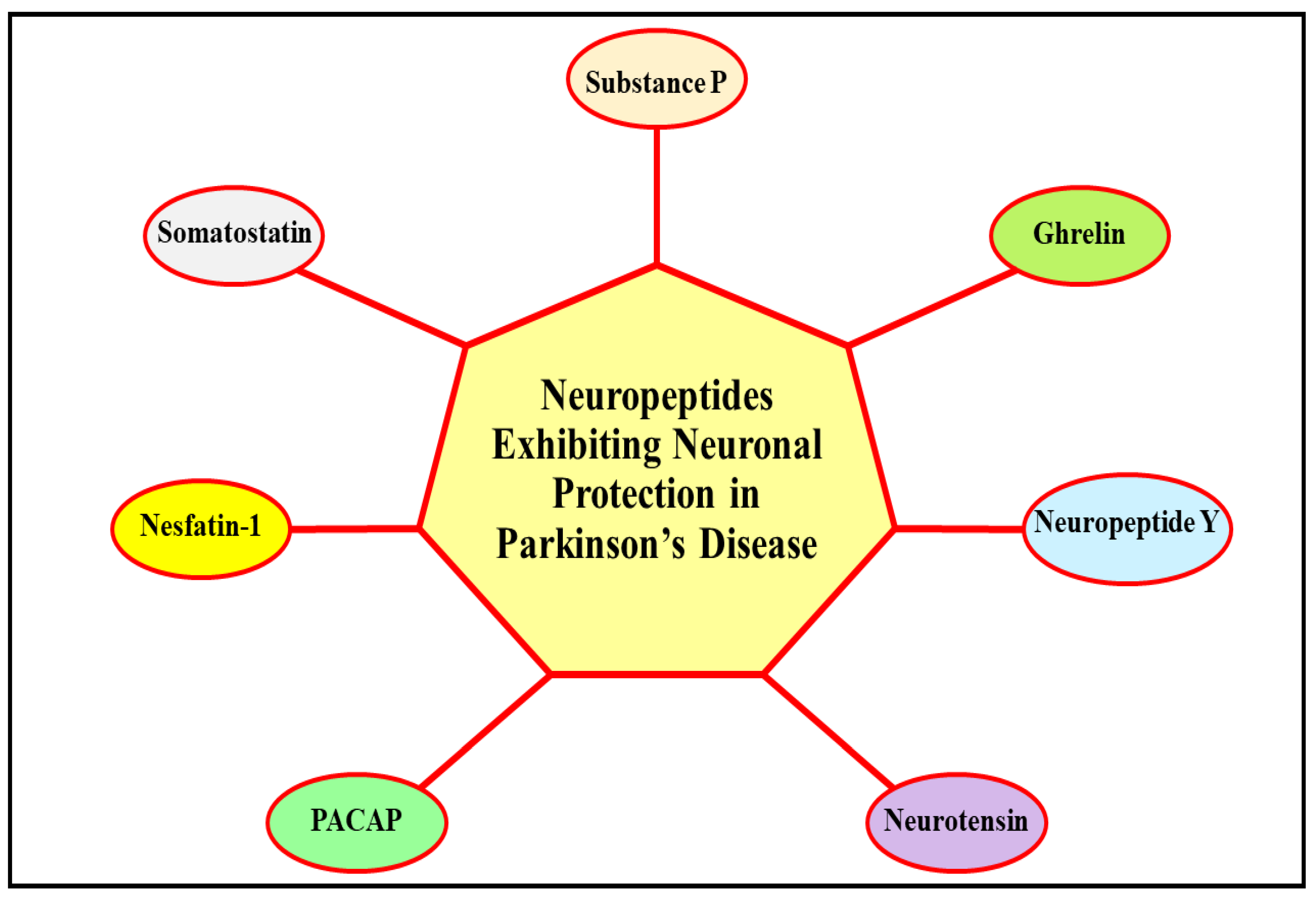
3. Deciphering the Neuroprotective Role of Neuropeptides in Parkinson’s Disease
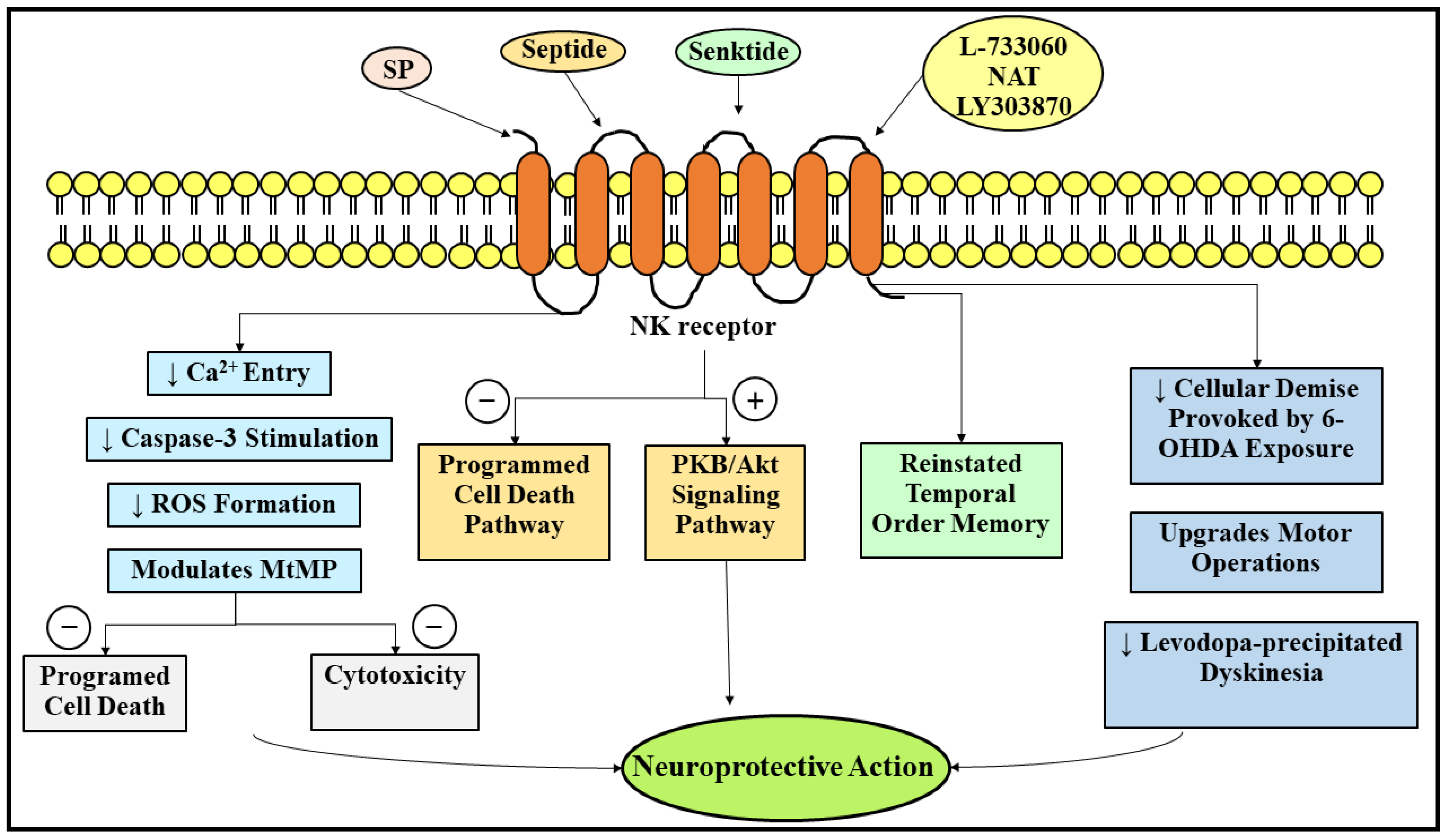
3.1. Neuroprotective Role of Substance P in Parkinson’s Disease
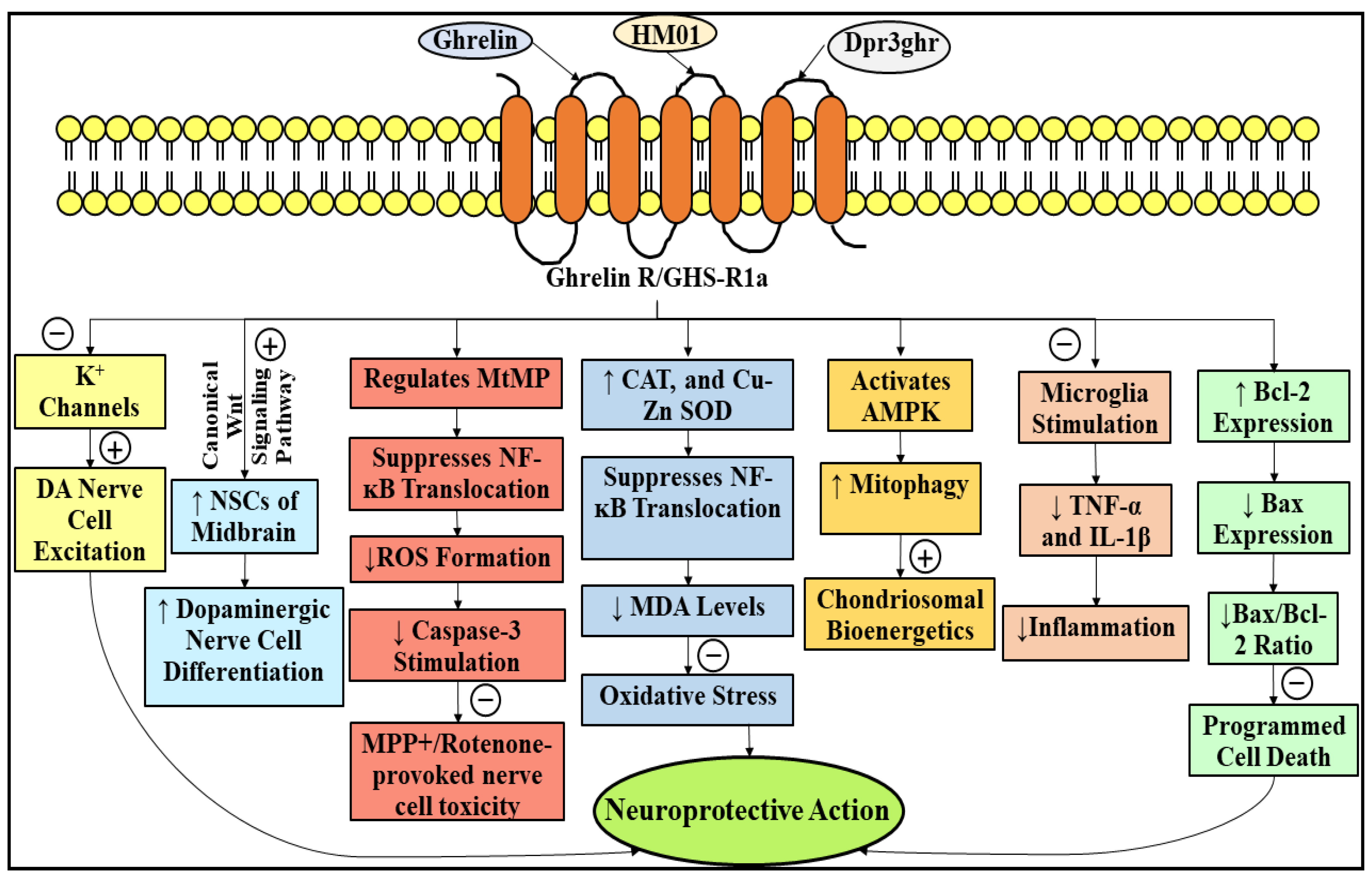
3.2. Neuroprotective Role of Ghrelin in Parkinson’s Disease
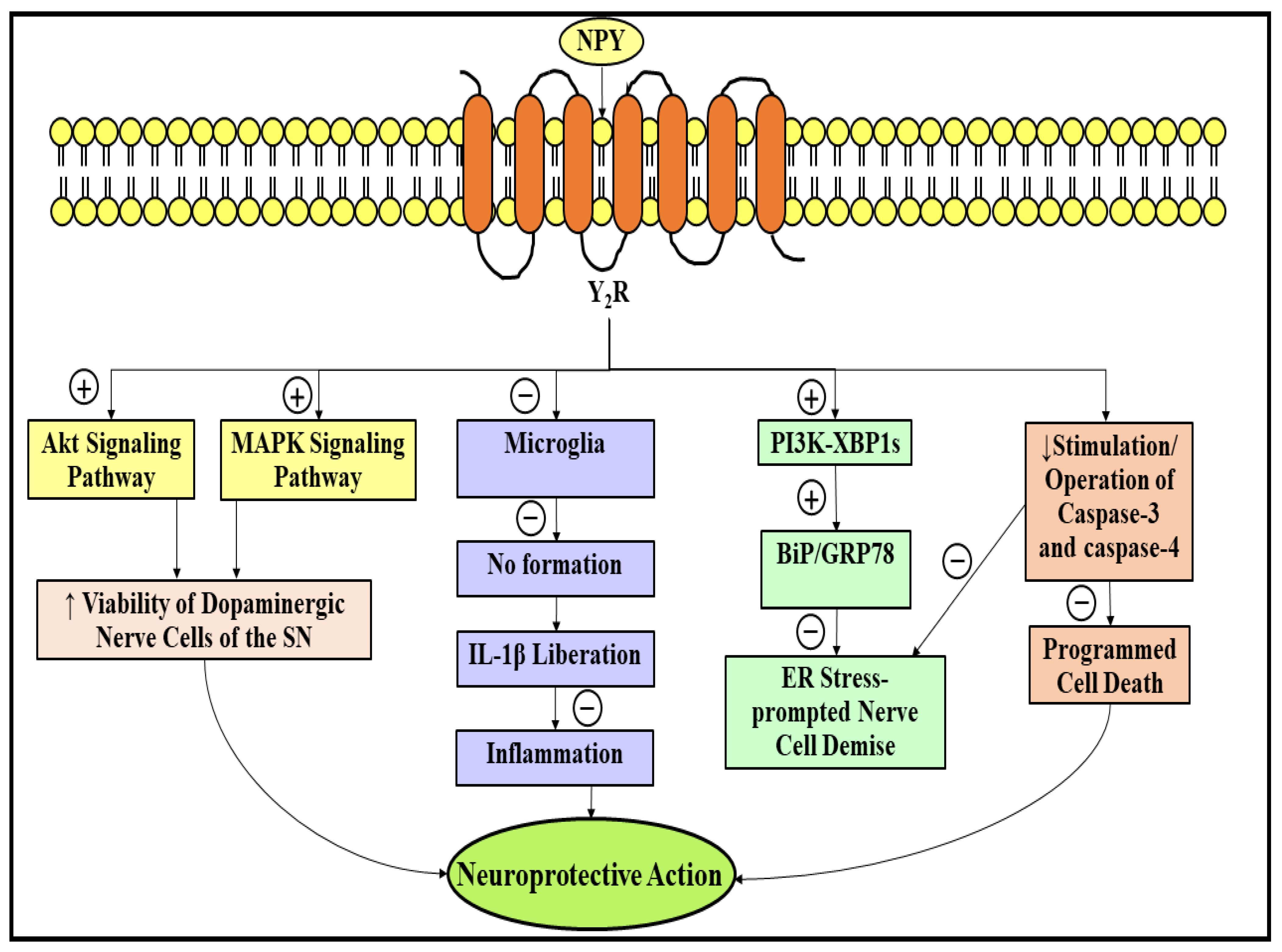
3.3. Neuroprotective Role of Neuropeptide Y in Parkinson’s Disease
3.4. Neurprotective Role of Neurotensin in Parkinson’s Disease
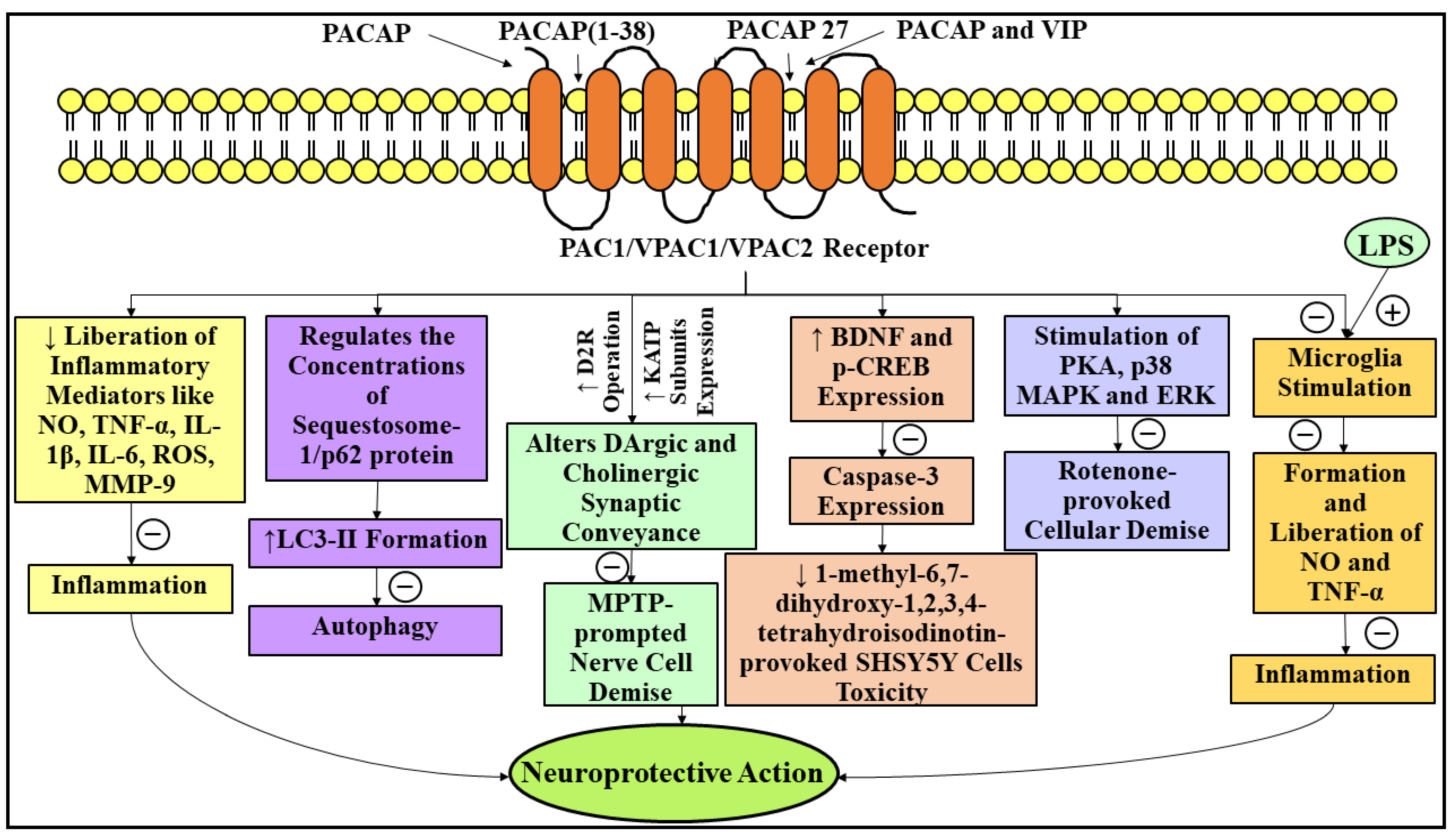
3.5. Neuroprotective Role of Pituitary Adenylate Cyclase-Activating Polypeptide in Parkinson’s Disease
3.6. Neuroprotective Role of Nesfatin-1 in Parkinson’s Disease
3.7. Neuroprotective Role of Somatostatin in Parkinson’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neuropeptide | Amino Acid Units | Amino Acid Sequence | Ref. |
|---|---|---|---|
| Substance P | 11 | H-Arg-Pro-Lys-Pro-Gln-Gln-Phe-Ple-Gly-Leu-Met-NH2 | [111,112] |
| Ghrelin | 28 | NH2-Gly-Ser-[Ser(n-octanoyl)]-Phe-Leu-Ser-Pro-Glu-His-Gln-Arg-Val-Gln-Gln-Arg-Lys-Glu-Ser-Lys-Lys-Pro-Pro-Ala-Lys-Leu-Gln-Pro-Arg-COOH | [131] |
| Neuropeptide Y | 36 | Tyr-Pro-Ser-Lys-Pro-Asp-Asn-Pro-Gly-Glu-Asp-Ala-Pro-Ala-Glu-Asp-Leu-Ala-Arg-Tyr-Tyr-Ser-Ala-Leu-Arg-His-Tyr-Ile-Asn-Leu-Ile-Thr-Arg-Gln-Arg-Tyr-NH2 | [156] |
| Neurotensin | 13 | pyr-Glu-Leu-Tyr-Glu-Asn-Lys-Pro-Arg-Arg-Pro-Tyr-Ile-Leu-OH | [177] |
| Pituitary adenylate cyclase-activating polypeptide | 27 | H-His-Ser-Asp-Gly-Ile-Phe-Thr-Asp-Ser-Tyr-Ser-Arg-Tyr-Arg-Lys-Gln-Met-Ala-Val-Lys-Lys-Tyr-Leu-Ala-Ala-Val-Leu-NH2 | [193,194] |
| 38 | H-His-Ser-Asp-Gly-Ile-Phe-Thr-Asp-Ser-Tyr-Ser-Arg-Tyr-Arg-Lys-Gln-Met-Ala-Val-Lys-Lys-Tyr-Leu-Ala-Ala-Val-Leu-Gly-Lys-Arg-Tyr-Lys-Gln-Arg-Val-Lys-Asn-Lys-NH2 | ||
| Nesfatin-1 | 82 | Val-Pro-Ile-Asp-Ile-Asp-Lys-Thr-Lys-Val-Gln-Asn-Ile-His-Pro-Val-Glu-Ser-Ala-Lys-Ile-Glu-Pro-Pro-Asp-Thr-Gly-Leu-Tyr-Tyr-Asp-Glu-Tyr-Leu-Lys-Gln-Val-Ile-Asp-Val-Leu-Glu-Thr-Asp-Lys-His-Phe-Arg-Glu-Lys-Leu-Gln-Lys-Ala-Asp-Ile-Glu-Glu-Ile-Lys-Ser-Gly-Arg-Leu-Ser-Lys-Glu-Leu-Asp-Leu-Val-Ser-His-His-Val-Arg-Thr-Lys-Leu-Asp-Glu-Leu | [218] |
| Somatostatin | 14 | Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys | [229] |
| 28 | Ser-Ala-Asn-Ser-Asn-Pro-Ala-Met-Ala-Pro-Arg-Glu-Arg-Lys-Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Palacios-Sánchez, L.; Nupan, M.T.; Botero-Meneses, J.S. James Parkinson and His Essay on “Shaking Palsy”, Two Hundred Years Later. Arq. Neuro-Psiquiatr. 2017, 75, 671–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M. Parkinson’s Disease: Clinical Manifestations and Treatment. Int. Rev. Psychiatry 2000, 12, 263–269. [Google Scholar] [CrossRef]
- Surmeier, D.J. Determinants of Dopaminergic Neuron Loss in Parkinson’s Disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Muangpaisan, W.; Hori, H.; Brayne, C. Systematic Review of the Prevalence and Incidence of Parkinson’s Disease in Asia. J. Epidemiol. 2009, 19, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.-M.; Liu, J.; Tian, Z.-Y.; Lu, D.; Zhou, Y.-Y. Systematic Review of the Prevalence and Incidence of Parkinson’s Disease in the People’s Republic of China. Neuropsychiatr. Dis. Treat. 2015, 11, 1467–1472. [Google Scholar] [CrossRef] [Green Version]
- Picillo, M.; Nicoletti, A.; Fetoni, V.; Garavaglia, B.; Barone, P.; Pellecchia, M.T. The Relevance of Gender in Parkinson’s Disease: A Review. J. Neurol. 2017, 264, 1583–1607. [Google Scholar] [CrossRef]
- Pang, S.Y.Y.; Ho, P.W.L.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The Interplay of Aging, Genetics and Environmental Factors in the Pathogenesis of Parkinson’s Disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Veldman, B.A.J.; Wijn, A.M.; Knoers, N.; Praamstra, P.; Horstink, M.W.I.M. Genetic and Environmental Risk Factors in Parkinson’s Disease. Clin. Neurol. Neurosurg. 1998, 100, 15–26. [Google Scholar] [CrossRef]
- Georgiou, A.; Demetriou, C.A.; Christou, Y.P.; Heraclides, A.; Leonidou, E.; Loukaides, P.; Yiasoumi, E.; Pantziaris, M.; Kleopa, K.A.; Papacostas, S.S.; et al. Genetic and Environmental Factors Contributing to Parkinson’s Disease: A Case-Control Study in the Cypriot Population. Front. Neurol. 2019, 10, 1047. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, F.; Zhang, S.; Xin, R.; Sun, Y. Genetic and Environmental Factors in Alzheimer’s and Parkinson’s Diseases and Promising Therapeutic Intervention via Fecal Microbiota Transplantation. NPJ Park. Dis. 2021, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Rachamalla, M.; Niyogi, S.; Datusalia, A.K.; Flora, S.J.S. Molecular Mechanism of Arsenic-Induced Neurotoxicity including Neuronal Dysfunctions. Int. J. Mol. Sci. 2021, 22, 10077. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.S.; Rachamalla, M.; Chary, N.R.; Shera, F.Y.; Tikoo, K.; Jena, G. Cytarabine Induced Cerebellar Neuronal Damage in Juvenile Rat: Correlating Neurobehavioral Performance with Cellular and Genetic Alterations. Toxicology 2012, 293, 41–52. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Kesari, K.K.; Rachamalla, M.; Mani, S.; Ashraf, G.M.; Jha, S.K.; Kumar, P.; Ambasta, R.K.; Dureja, H.; Devkota, H.P.; et al. CRISPR/Cas9 Gene Editing: New Hope for Alzheimer’s Disease Therapeutics. J. Adv. Res. 2021. In press. [Google Scholar] [CrossRef]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Iarkov, A.; Barreto, G.E.; Grizzell, J.A.; Echeverria, V. Strategies for the Treatment of Parkinson’s Disease: Beyond Dopamine. Front. Aging Neurosci. 2020, 12, 4. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA—J. Am. Med. Assoc. 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Kakkar, A.K.; Dahiya, N. Management of Parkinsonߣs Disease: Current and Future Pharmacotherapy. Eur. J. Pharmacol. 2015, 750, 74–81. [Google Scholar] [CrossRef]
- Carniglia, L.; Ramírez, D.; Durand, D.; Saba, J.; Turati, J.; Caruso, C.; Scimonelli, T.N.; Lasaga, M. Neuropeptides and Microglial Activation in Inflammation, Pain, and Neurodegenerative Diseases. Mediat. Inflamm. 2017, 2017, 5048616. [Google Scholar] [CrossRef]
- Hökfelt, T.; Barde, S.; Xu, Z.-Q.D.; Kuteeva, E.; Rüegg, J.; Le Maitre, E.; Risling, M.; Kehr, J.; Ihnatko, R.; Theodorsson, E.; et al. Neuropeptide and Small Transmitter Coexistence: Fundamental Studies and Relevance to Mental Illness. Front. Neural Circuits 2018, 12, 106. [Google Scholar] [CrossRef] [Green Version]
- Merighi, A. Costorage and Coexistence of Neuropeptides in the Mammalian CNS. Prog. Neurobiol. 2002, 66, 161–190. [Google Scholar] [CrossRef]
- Merighi, A.; Salio, C.; Ferrini, F.; Lossi, L. Neuromodulatory Function of Neuropeptides in the Normal CNS. J. Chem. Neuroanat. 2011, 42, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Surget, A.; Leman, S.; Griebel, G.; Belzung, C.; Yalcin, I. Neuropeptides in Psychiatric Diseases: An Overview with a Particular Focus on Depression and Anxiety Disorders. CNS Neurol. Disord.—Drug Targets 2006, 5, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, A.J. Proteases. Curr. Protoc. Protein Sci. 2000, 21, 21.1.1–21.1.12. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R. Cell-Surface Peptidases. Int. Rev. Cytol. 2004, 235, 165–213. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Parks, G.S.; Civelli, O. Novel Neuropeptides as Ligands of Orphan G Protein-Coupled Receptors. Curr. Pharm. Des. 2012, 17, 2626–2631. [Google Scholar] [CrossRef] [Green Version]
- Hökfelt, T.; Bartfai, T.; Bloom, F. Neuropeptides: Opportunities for Drug Discovery. Lancet Neurol. 2003, 2, 463–472. [Google Scholar] [CrossRef]
- Palkovits, M. Neuropeptide Messenger Plasticity in the CNS Neurons Following Axotomy. Mol. Neurobiol. 1995, 10, 91–103. [Google Scholar] [CrossRef]
- Catalani, E.; De Palma, C.; Perrotta, C.; Cervia, D. Current Evidence for a Role of Neuropeptides in the Regulation of Autophagy. BioMed Res. Int. 2017, 2017, 5856071. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Chen, L.; Xue, Y.; Xia, Y.-J. Substance P Prevents 1-methyl-4-Phenylpyridiniuminduced Cytotoxicity through Inhibition of Apoptosis via Neurokinin-1 Receptors in MES23.5 Cells. Mol. Med. Rep. 2012, 12, 8085–8092. [Google Scholar] [CrossRef]
- Shi, L.; Du, X.; Jiang, H.; Xie, J. Ghrelin and Neurodegenerative Disorders—A Review. Mol. Neurobiol. 2017, 54, 1144–1155. [Google Scholar] [CrossRef]
- Bayliss, J.A.; Lemus, M.; Santos, V.V.; Deo, M.; Elsworth, J.D.; Andrews, Z.B. Acylated but not Des-acyl Ghrelin is Neuroprotective in an MPTP Mouse Model of Parkinson’s Disease. J. Neurochem. 2016, 137, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, X.; Liu, S.; Zhao, Y.; Zhu, J.; Liu, K. Roles of Neuropeptide Y in Neurodegenerative and Neuroimmune Diseases. Front. Neurosci. 2019, 13, 869. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, M.; Popatanasov, A.; Klissurov, R.; Stoeva, S.; Pajpanova, T.; Kalfin, R.; Tancheva, L. Preventive Effect of Two New Neurotensin Analogues on Parkinson’s Disease Rat Model. J. Mol. Neurosci. 2018, 66, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Maasz, G.; Zrinyi, Z.; Reglodi, D.; Petrovics, D.; Rivnyak, A.; Kiss, T.; Jungling, A.; Tamas, A.; Pirger, Z. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Has Neuroprotective Function in Dopamine-Based Neurodegeneration Developed in Two Parkinsonian Models. Dis. Model. Mech. 2017, 10, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Xie, J.; Wang, J. Neuroprotective Effects of Brain-Gut Peptides: A Potential Therapy for Parkinson’s Disease. Neurosci. Bull. 2019, 35, 1085–1096. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, X.; Li, X.; Liu, N.; Lou, F.; Ma, H.; Luo, X.; Ren, Y. Somatostatin Prevents Lipopolysaccharide-Induced Neurodegeneration in the Rat Substantia Nigra by Inhibiting the Activation of Microglia. Mol. Med. Rep. 2015, 12, 1002–1008. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, E.; Chandrasekhar, G.; Chandrasekhar, P.; Anbarasu, K.; Vickram, A.S.; Karunakaran, R.; Rajasekaran, R.; Srikumar, P.S. Alpha-Synuclein Aggregation in Parkinson’s Disease. Front. Med. 2021, 8, 736978. [Google Scholar] [CrossRef]
- Coelho, M.; Ferreira, J.; Rosa, M.; Sampaio, C. Treatment Options for Non-motor Symptoms in Late-Stage Parkinson’s Disease. Expert Opin. Pharmacother. 2008, 9, 523–535. [Google Scholar] [CrossRef]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Dorsey, E.R.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and Projected Future Economic Burden of Parkinson’s Disease in the U.S. NPJ Park. Dis. 2020, 6, 15. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A.; Logroscino, G.; Gaziano, J.M.; Kurth, T. Incidence and Remaining Lifetime Risk of Parkinson Disease in Advanced Age. Neurology 2009, 72, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Crispino, P.; Gino, M.; Barbagelata, E.; Ciarambino, T.; Politi, C.; Ambrosino, I.; Ragusa, R.; Marranzano, M.; Biondi, A.; Vacante, M. Gender Differences and Quality of Life in Parkinson’s Disease. Int. J. Environ. Res. Public Health 2021, 18, 198. [Google Scholar] [CrossRef]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Parkinson’s Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current Understanding of the Molecular Mechanisms in Parkinson’s Disease: Targets for Potential Treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Koros, C.; Strohäker, T.; Schulte, C.; Bozi, M.; Varvaresos, S.; Ibáñez de Opakua, A.; Simitsi, A.M.; Bougea, A.; Voumvourakis, K.; et al. A Novel SNCA A30G Mutation Causes Familial Parkinson’s Disease. Mov. Disord. 2021, 36, 1624–1633. [Google Scholar] [CrossRef]
- Magistrelli, L.; Contaldi, E.; Comi, C. The Impact of SNCA Variations and Its Product Alpha-Synuclein on Non-Motor Features of Parkinson’s Disease. Life 2021, 11, 804. [Google Scholar] [CrossRef]
- Kamienieva, I.; Duszyński, J.; Szczepanowska, J. Multitasking Guardian of Mitochondrial Quality: Parkin Function and Parkinson’s Disease. Transl. Neurodegener. 2021, 10, 5. [Google Scholar] [CrossRef]
- Nawaz, M.S.; Asghar, R.; Pervaiz, N.; Ali, S.; Hussain, I.; Xing, P.; Bao, Y.; Abbasi, A.A. Molecular Evolutionary and Structural Analysis of Human UCHL1 Gene Demonstrates the Relevant Role of Intragenic Epistasis in Parkinson’s Disease and Other Neurological Disorders. BMC Evol. Biol. 2020, 20, 130. [Google Scholar] [CrossRef]
- Vizziello, M.; Borellini, L.; Franco, G.; Ardolino, G. Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease. Cells 2021, 10, 3022. [Google Scholar] [CrossRef]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, E.-M.; Lee, B.D. LRRK2 at the Crossroad of Aging and Parkinson’s Disease. Genes 2021, 12, 505. [Google Scholar] [CrossRef]
- Cerri, S.; Ghezzi, C.; Ongari, G.; Croce, S.; Avenali, M.; Zangaglia, R.; Di Monte, D.A.; Valente, E.M.; Blandini, F. GBA Mutations Influence the Release and Pathological Effects of Small Extracellular Vesicles from Fibroblasts of Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 2215. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Xu, Z.; Liao, S. VPS35, the Core Component of the Retromer Complex, and Parkinson’s Disease. Ibrain 2021, 7, 318–324. [Google Scholar] [CrossRef]
- Park, J.-S.; Blair, N.F.; Sue, C.M. The Role of ATP13A2 in Parkinson’s Disease: Clinical Phenotypes and Molecular Mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-C.; Huang, P.; Li, Q.-Q.; Sun, Q.; Li, D.-H.; Wang, T.; Shen, J.-Y.; Du, J.-J.; Cui, S.-S.; Gao, C.; et al. Mutation Analysis ofHTRA2Gene in Chinese Familial Essential Tremor and Familial Parkinson’s Disease. Park. Dis. 2017, 2017, 3217474. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Mangone, G.; Tesson, C.; Bertrand, H.; Benmahdjoub, M.; Kesraoui, S.; Arezki, M.; Singleton, A.; Corvol, J.-C.; Brice, A. Clinical Variability of SYNJ1-Associated Early-Onset Parkinsonism. Front. Neurol. 2021, 12, 648457. [Google Scholar] [CrossRef]
- Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Chigurupati, S.; Alrashdi, I.; Bungau, S.G. Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target. Int. J. Mol. Sci. 2021, 22, 10161. [Google Scholar] [CrossRef]
- Thirugnanam, T.; Santhakumar, K. Chemically Induced Models of Parkinson’s Disease. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2022, 252, 109213. [Google Scholar] [CrossRef]
- Kochmanski, J.; VanOeveren, S.E.; Patterson, J.R.; Bernstein, A.I. Developmental Dieldrin Exposure Alters DNA Methylation at Genes Related to Dopaminergic Neuron Development and Parkinson’s Disease in Mouse Midbrain. Toxicol. Sci. 2019, 169, 593–607. [Google Scholar] [CrossRef]
- Jia, Z.; Misra, H.P. Exposure to Mixtures of Endosulfan and Zineb Induces Apoptotic and Necrotic Cell Death in SH-SY5Y Neuroblastoma Cells, In Vitro. J. Appl. Toxicol. 2007, 27, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.A.; Myers, K.M.; Chen, A.; Martin, N.T.; Barajas, A.; Schweizer, F.E.; Krantz, D.E. Ziram, a Pesticide Associated with Increased Risk for Parkinson’s Disease, Differentially Affects the Presynaptic Function of Aminergic and Glutamatergic Nerve Terminals at the Drosophila Neuromuscular Junction. Exp. Neurol. 2016, 275, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouchieu, C.; Piel, C.; Carles, C.; Gruber, A.; Helmer, C.; Tual, S.; Marcotullio, E.; LeBailly, P.; Baldi, I. Pesticide Use in Agriculture and Parkinson’s Disease in the AGRICAN Cohort Study. Int. J. Epidemiol. 2018, 47, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.C.; Marentette, J.O.; Rauniyar, A.K.; Prutton, K.M.; Khatri, M.; Matheson, C.; Reisz, J.A.; Reigan, P.; D’Alessandro, A.; Roede, J.R. Maneb Alters Central Carbon Metabolism and Thiol Redox Status in a Toxicant Model of Parkinson’s Disease. Free Radic. Biol. Med. 2021, 162, 65–76. [Google Scholar] [CrossRef]
- Lock, E.A.; Zhang, J.; Checkoway, H. Solvents and Parkinson Disease: A Systematic Review of Toxicological and Epidemiological Evidence. Toxicol. Appl. Pharmacol. 2013, 266, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Goldman, S.M.; Quinlan, P.J.; Ross, G.W.; Marras, C.; Meng, C.; Bhudhikanok, G.S.; Comyns, K.; Korell, M.; Chade, A.R.; Kasten, M.; et al. Solvent Exposures and Parkinson Disease Risk in Twins. Ann. Neurol. 2012, 71, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Anwer, K.; Makeen, H.A.; Albratty, M.; Mohan, S.; Bungau, S. Mechanistic Insights Expatiating the Redox-Active-Metal-Mediated Neuronal Degeneration in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 678. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective Neuronal Vulnerability to Oxidative Stress in the Brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I Deficiency in Parkinson’s Disease Frontal Cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.F.; Harris, T.A. Dopamine and Uric Acid Act as Antioxidants in the Repair of DNA Radicals: Implications in Parkinson’s Disease. Free Radic. Res. 2003, 37, 1131–1136. [Google Scholar] [CrossRef]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s Disease: Dopamine, Vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Zhuang, Q.-Q.; Zhu, L.-B.; Zhu, H.; Li, T.; Li, R.; Chen, S.-F.; Huang, C.-P.; Zhang, X.; Zhu, J.-H. Meta-Analysis of Brain Iron Levels of Parkinson’s Disease Patients Determined by Postmortem and MRI Measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar] [CrossRef] [Green Version]
- De Farias, C.C.; Maes, M.; Bonifácio, K.L.; Bortolasci, C.C.; de Souza Nogueira, A.; Brinholi, F.F.; Matsumoto, A.K.; do Nascimento, M.A.; De Melo, L.B.; Nixdorf, S.L.; et al. Highly Specific Changes in Antioxidant Levels and Lipid Peroxidation in Parkinson’s Disease and Its Progression: Disease and Staging Biomarkers and New Drug Targets. Neurosci. Lett. 2016, 617, 66–71. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.; Alonso-Navarro, H.; Herrero, M.; García-Martín, E.; Agúndez, J. An Update on the Role of Nitric Oxide in the Neurodegenerative Processes of Parkinson’s Disease. Curr. Med. Chem. 2016, 23, 2666–2679. [Google Scholar] [CrossRef]
- Subedi, L.; Gaire, B.; Parveen, A.; Kim, S.-Y. Nitric Oxide as a Target for Phytochemicals in Anti-Neuroinflammatory Prevention Therapy. Int. J. Mol. Sci. 2021, 22, 4771. [Google Scholar] [CrossRef]
- Senkevich, K.; Gan-Or, Z. Autophagy Lysosomal Pathway Dysfunction in Parkinson’s Disease; Evidence from Human Genetics. Park. Relat. Disord. 2020, 73, 60–71. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The Role of Autophagy in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beilina, A.; Cookson, M.R. Genes Associated with Parkinson’s Disease: Regulation of Autophagy and Beyond. J. Neurochem. 2016, 139, 91–107. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal Function Is Impaired in Substantia Nigra in Parkinson’s Disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. Rare Genetic Mutations Shed Light on the Pathogenesis of Parkinson Disease. J. Clin. Investig. 2003, 111, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 Promotes Atypical Ubiquitination of Mutant DJ-1 and Alpha-Synuclein and Is Localized to Lewy Bodies in Sporadic Parkinson’s Disease Brains. Hum. Mol. Genet. 2010, 19, 3759–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaught, K.S.P.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered Proteasomal Function in Sporadic Parkinson’s Disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2021, 8, 615461. [Google Scholar] [CrossRef]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Bose, A.; Beal, M.F. Mitochondrial Dysfunction in Parkinson’s Disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, Paraquat, and Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaerts, V.; Theuns, J.; Van Broeckhoven, C. Genetic Findings in Parkinson’s Disease and Translation into Treatment: A Leading Role for Mitochondria? Genes Brain Behav. 2008, 7, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and Autophagy in Nigral Neurons of Patients with Parkinson’s Disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.-P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear Translocation of NF-κB is Increased in Dopaminergic Neurons of Patients with Parkinson Disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular Mechanisms of Cell Death in Neurological Diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Mount, M.P.; Lira, A.; Grimes, D.; Smith, P.D.; Faucher, S.; Slack, R.; Anisman, H.; Hayley, S.; Park, D.S. Involvement of Interferon-γ in Microglial-Mediated Loss of Dopaminergic Neurons. J. Neurosci. 2007, 27, 3328–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellucci, A.; Bubacco, L.; Longhena, F.; Parrella, E.; Faustini, G.; Porrini, V.; Bono, F.; Missale, C.; Pizzi, M. Nuclear Factor-κB Dysregulation and α-Synuclein Pathology: Critical Interplay in the Pathogenesis of Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, A.; Mouatt-Prigent, A.; Vila, M.; Abbas, N.; Perier, C.; Faucheux, B.A.; Vyasb, S.; Hirsch, E.C. Increased Expression and Redistribution of the Antiapoptotic Molecule Bcl-xL in Parkinson’s Disease. Neurobiol. Dis. 2002, 10, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Badanjak, K.; Fixemer, S.; Smajić, S.; Skupin, A.; Grünewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein Activates Microglia: A Process Leading to Disease Progression in Parkinson’s Disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- He, S.; Zhong, S.; Liu, G.; Yang, J. Alpha-Synuclein: The Interplay of Pathology, Neuroinflammation, and Environmental Factors in Parkinson’s Disease. Neurodegener. Dis. 2021, 20, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.-H.; Castillo, P.; Shinsky, N.; García-Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice Lacking α-Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated In Vitro Fibril Formation by a Mutant α-synuclein Linked to Early-Onset Parkinson Disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, E.A.; Graves, C.L.; Mishizen-Eberz, A.J.; Lupoli, M.A.; Lynch, D.R.; Englander, S.W.; Axelsen, P.H.; Giasson, B.I. The E46K Mutation in α-Synuclein Increases Amyloid Fibril Formation. J. Biol. Chem. 2005, 280, 7800–7807. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M.-Y. Addition of Exogenous α-Synuclein Preformed Fibrils to Primary Neuronal Cultures to Seed Recruitment of Endogenous α-Synuclein to Lewy Body and Lewy Neurite–like Aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-Synuclein Fibrils Seed the Formation of Lewy Body-like Intracellular Inclusions in Cultured Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, F.; Abeywardana, T.; Dhall, A.; Marotta, N.P.; Varkey, J.; Langen, R.; Chatterjee, C.; Pratt, M.R. Semisynthetic, Site-Specific Ubiquitin Modification of α-Synuclein Reveals Differential Effects on Aggregation. J. Am. Chem. Soc. 2012, 134, 5468–5471. [Google Scholar] [CrossRef] [Green Version]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M.Y. Lewy Body-like α-Synuclein Aggregates Resist Degradation and Impair Macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [Green Version]
- Euler, U.S.V.; Gaddum, J.H. An Unidentified Depressor Substance in Certain Tissue Extracts. J. Physiol. 1931, 72, 74–87. [Google Scholar] [CrossRef]
- Chang, M.M.; Leeman, S.E.; Niall, H.D. Amino-acid Sequence of Substance P. Nat. New Biol. 1971, 232, 86–87. [Google Scholar] [CrossRef]
- Nicoll, R.A.; Schenker, C.; Leeman, S.E. Substance P as a Transmitter Candidate. Annu. Rev. Neurosci. 1980, 3, 227–268. [Google Scholar] [CrossRef] [PubMed]
- Warden, M.K.; Young, W.S. Distribution of Cells Containing mRNAs Encoding Substance P and Neurokinin B in the Rat Central Nervous System. J. Comp. Neurol. 1988, 272, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide Substance P and the Immune Response. Cell Mol. Life Sci. 2016, 73, 4249–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, T.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Abdeen, A.; Ibrahim, S.F.; Mani, V.; Iqbal, M.S.; Bhatia, S.; et al. Exploring the Role of Neuropeptides in Depression and Anxiety. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2022, 114, 110478. [Google Scholar] [CrossRef]
- Mauborgne, A.; Javoy-Agid, F.; Legrand, J.C.; Agid, Y.; Cesselin, F. Decrease of Substance P-like Immunoreactivity in the Substantia Nigra and Pallidum of Parkinsonian Brains. Brain Res. 1983, 268, 167–170. [Google Scholar] [CrossRef]
- Tenovuo, O.; Rinne, U.K.; Viljanen, M.K. Substance P Immunoreactivity in the Post-mortem Parkinsonian Brain. Brain Res. 1984, 303, 113–116. [Google Scholar] [CrossRef]
- Schröder, J.B.; Marian, T.; Claus, I.; Muhle, P.; Pawlowski, M.; Wiendl, H.; Suntrup-Krueger, S.; Meuth, S.G.; Dziewas, R.; Ruck, T.; et al. Substance P Saliva Reduction Predicts Pharyngeal Dysphagia in Parkinson’s Disease. Front. Neurol. 2019, 10, 386. [Google Scholar] [CrossRef]
- Lindefors, N.; Brodin, E.; Tossman, U.; Segovia, J.; Ungerstedt, U. Tissue Levels and in Vivo Release of Tachykinins and GABA in Striatum and Substantia Nigra of Rat Brain after Unilateral Striatal Dopamine Denervation. Exp. Brain Res. 1989, 74, 527–534. [Google Scholar] [CrossRef]
- Thornton, E.; Vink, R. Treatment with a Substance P Receptor Antagonist Is Neuroprotective in the Intrastriatal 6-Hydroxydopamine Model of Early Parkinson’s Disease. PLoS ONE 2012, 7, e34138. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhang, L.; Xie, J.; Shi, L. The Emerging Role of Neuropeptides in Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 90. [Google Scholar] [CrossRef]
- Thornton, E.; Tran, T.T.B.; Vink, R. A Substance P Mediated Pathway Contributes to 6-hydroxydopamine Induced Cell Death. Neurosci. Lett. 2010, 481, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.M.T.; Chen, L.W.; Chan, Y.S.; Yung, K.K.L. Neuroprotective Effects of Neurokinin Receptor One in Dopaminergic Neurons Are Mediated through Akt/PKB Cell Signaling Pathway. Neuropharmacology 2011, 61, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Pieri, M.; Ciotti, M.T.; Carunchio, I.; Canu, N.; Calissano, P.; Zona, C.; Severini, C. Substance P Provides Neuroprotection in Cerebellar Granule Cells through Akt and MAPK/Erk Activation: Evidence for the Involvement of the Delayed Rectifier Potassium Current. Neuropharmacology 2007, 52, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Chao, O.Y.; Wang, A.-L.; Nikolaus, S.; De Souza Silva, M.A. NK3 Receptor Agonism Reinstates Temporal Order Memory in the Hemiparkinsonian Rat. Behav. Brain Res. 2015, 285, 208–212. [Google Scholar] [CrossRef]
- Thornton, E.; Hassall, M.M.; Corrigan, F.; Vink, R. The NK1 Receptor Antagonist N-acetyl-l-tryptophan Reduces Dyskinesia in a Hemi-parkinsonian Rodent Model. Park. Relat. Disord. 2014, 20, 508–513. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, H.; Shi, H.; Wang, X.; Zhang, S.; Zhang, Z.; Zu, J.; Zhang, W.; Shen, X.; Cui, G.; et al. Intranigral Administration of Substance P Receptor Antagonist Attenuated Levodopa-Induced Dyskinesia in a Rat Model of Parkinson’s Disease. Exp. Neurol. 2015, 271, 168–174. [Google Scholar] [CrossRef]
- Kim, W.-G.; Mohney, R.P.; Wilson, B.; Jeohn, G.-H.; Liu, B.; Hong, J.-S. Regional Difference in Susceptibility to Lipopolysaccharide-Induced Neurotoxicity in the Rat Brain: Role of Microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin Is a Growth-Hormone-Releasing Acylated Peptide from Stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Serrenho, D.; Santos, S.D.; Carvalho, A.L. The Role of Ghrelin in Regulating Synaptic Function and Plasticity of Feeding-Associated Circuits. Front. Cell. Neurosci. 2019, 13, 205. [Google Scholar] [CrossRef]
- St-Pierre, D.H.; Wang, L.; Taché, Y. Ghrelin: A Novel Player in the Gut-Brain Regulation of Growth Hormone and Energy Balance. Physiology 2003, 18, 242–246. [Google Scholar] [CrossRef]
- Frago, L.M.; Ebaquedano, E.; Eargente, J.; Chowen, J.A. Neuroprotective Actions of Ghrelin and Growth Hormone Secretagogues. Front. Mol. Neurosci. 2011, 4, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, D.; Johnson, A.L.; Lelos, M.; Smith, G.; Roberts, L.D.; Phelps, L.; Dunnett, S.B.; Morgan, A.H.; Brown, R.M.; Wells, T.; et al. Acyl-Ghrelin Attenuates Neurochemical and Motor Deficits in the 6-OHDA Model of Parkinson’s Disease. bioRxiv 2022. [Google Scholar] [CrossRef]
- He, X.; Yuan, W.; Liu, F.; Feng, J.; Guo, Y. Acylated Ghrelin is Protective Against 6-OHDA-induced Neurotoxicity by Regulating Autophagic Flux. Front. Pharmacol. 2021, 11, 586302. [Google Scholar] [CrossRef]
- Wagner, J.; Vulinović, F.; Grünewald, A.; Unger, M.M.; Möller, J.C.; Klein, C.; Michel, P.P.; Ries, V.; Oertel, W.H.; Alvarez-Fischer, D. Acylated and Unacylated Ghrelin Confer Neuroprotection to Mesencephalic Neurons. Neuroscience 2017, 365, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Wang, W.; Jia, F.; Du, X.; Xie, A.; He, Q.; Shen, X.; Zhang, J.; Rogers, J.; Xie, J.; et al. Assessments of Plasma Ghrelin Levels in the Early Stages of Parkinson’s Disease. Mov. Disord. 2017, 32, 1487–1491. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Erion, D.; Beiler, R.; Liu, Z.-W.; Abizaid, A.; Zigman, J.; Elsworth, J.D.; Savitt, J.M.; DiMarchi, R.; Tschoep, M.; et al. Ghrelin Promotes and Protects Nigrostriatal Dopamine Function via a UCP2-Dependent Mitochondrial Mechanism. J. Neurosci. 2009, 29, 14057–14065. [Google Scholar] [CrossRef] [PubMed]
- Suda, Y.; Kuzumaki, N.; Sone, T.; Narita, M.; Tanaka, K.; Hamada, Y.; Iwasawa, C.; Shibasaki, M.; Maekawa, A.; Matsuo, M.; et al. Down-Regulation of Ghrelin Receptors on Dopaminergic Neurons in the Substantia Nigra Contributes to Parkinson’s Disease-like Motor Dysfunction. Mol. Brain 2018, 11, 6. [Google Scholar] [CrossRef]
- Jiang, H.; Li, L.-J.; Wang, J.; Xie, J.-X. Ghrelin Antagonizes MPTP-induced Neurotoxicity to the Dopaminergic Neurons in Mouse Substantia Nigra. Exp. Neurol. 2008, 212, 532–537. [Google Scholar] [CrossRef]
- Wang, H.; Dou, S.; Zhu, J.; Shao, Z.; Wang, C.; Cheng, B. Ghrelin Protects Dopaminergic Neurons against MPTP Neurotoxicity through Promoting Autophagy and Inhibiting Endoplasmic Reticulum Mediated Apoptosis. Brain Res. 2020, 1746, 147023. [Google Scholar] [CrossRef]
- Moon, M.; Kim, H.G.; Hwang, L.; Seo, J.-H.; Kim, S.; Hwang, S.; Kim, S.; Lee, D.; Chung, H.; Oh, M.S.; et al. Neuroprotective Effect of Ghrelin in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson’s Disease by Blocking Microglial Activation. Neurotox. Res. 2009, 15, 332–347. [Google Scholar] [CrossRef]
- Yu, J.; Xu, H.; Shen, X.; Jiang, H. Ghrelin Protects MES23.5 Cells against Rotenone via Inhibiting Mitochondrial Dysfunction and Apoptosis. Neuropeptides 2016, 56, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, S.; Ren, J.; Li, B.; Qin, B. Ghrelin Protects Retinal Ganglion Cells against Rotenone via Inhibiting Apoptosis, Restoring Mitochondrial Function, and Activating AKT-mTOR Signaling. Neuropeptides 2018, 67, 63–70. [Google Scholar] [CrossRef]
- Bayliss, J.A.; Lemus, M.B.; Stark, R.; Santos, V.V.; Thompson, A.; Rees, D.J.; Galic, S.; Elsworth, J.D.; Kemp, B.E.; Davies, J.S.; et al. Ghrelin-AMPK Signaling Mediates the Neuroprotective Effects of Calorie Restriction in Parkinson’s Disease. J. Neurosci. 2016, 36, 3049–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Bian, X.; Qu, Z.; Ma, Z.; Zhou, Y.; Wang, K.; Jiang, H.; Xie, J. Peptide Hormone Ghrelin Enhances Neuronal Excitability by Inhibition of Kv7/KCNQ Channels. Nat. Commun. 2013, 4, 1435. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Ma, Z.; Shi, L.; Xie, J. Effects of Ghrelin on the Electrical Activities of Substantia Nigra Dopaminergic Neurons Treated with MPP+. Neurochem. Int. 2020, 138, 104780. [Google Scholar] [CrossRef]
- Gong, B.; Jiao, L.; Du, X.; Li, Y.; Bi, M.; Jiao, Q.; Jiang, H. Ghrelin Promotes Midbrain Neural Stem Cells Differentiation to Dopaminergic Neurons through Wnt/β-catenin Pathway. J. Cell. Physiol. 2020, 235, 8558–8570. [Google Scholar] [CrossRef] [PubMed]
- Minalyan, A.; Gabrielyan, L.; Pietra, C.; Taché, Y.; Wang, L. Multiple Beneficial Effects of Ghrelin Agonist, HM01 on Homeostasis Alterations in 6-Hydroxydopamine Model of Parkinson’s Disease in Male Rats. Front. Integr. Neurosci. 2019, 13, 13. [Google Scholar] [CrossRef]
- Popelová, A.; Kákonová, A.; Hrubá, L.; Kunes, J.; Maletínská, L.; Železná, B. Potential Neuroprotective and Anti-Apoptotic Properties of a Long-Lasting Stable Analog of Ghrelin: An In Vitro Study Using SH-SY5Y Cells. Physiol. Res. 2018, 67, 339–346. [Google Scholar] [CrossRef]
- Morgan, A.H.; Rees, D.J.; Andrews, Z.B.; Davies, J.S. Ghrelin Mediated Neuroprotection—A Possible Therapy for Parkinson’s Disease? Neuropharmacology 2018, 136, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Song, N.; Xie, J.; Jiang, H. Ghrelin Antagonized 1-Methyl-4-Phenylpyridinium (MPP+)-Induced Apoptosis in MES23.5 Cells. J. Mol. Neurosci. 2009, 37, 182–189. [Google Scholar] [CrossRef]
- Liu, L.; Xu, H.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Ghrelin Prevents 1-methyl-4-phenylpyridinium Ion-induced Cytotoxicity through Antioxidation and NF-κB Modulation in MES23.5 cells. Exp. Neurol. 2010, 222, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, J.A.; Andrews, Z.B. Ghrelin Is Neuroprotective in Parkinson’s Disease: Molecular Mechanisms of Metabolic Neuroprotection. Ther. Adv. Endocrinol. Metab. 2013, 4, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatemoto, K.; Carlquist, M.; Mutt, V. Neuropeptide Y—A Novel Brain Peptide with Structural Similarities to Peptide YY and Pancreatic Polypeptide. Nature 1982, 296, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Gehlert, D.R. Introduction to the Reviews on Neuropeptide Y. Neuropeptides 2004, 38, 135–140. [Google Scholar] [CrossRef]
- Tatemoto, K. Neuropeptide Y: Complete Aamino Acid Sequence of the Brain Peptide. Proc. Natl. Acad. Sci. USA 1982, 79, 5485–5489. [Google Scholar] [CrossRef] [Green Version]
- Shiozaki, K.; Kawabe, M.; Karasuyama, K.; Kurachi, T.; Hayashi, A.; Ataka, K.; Iwai, H.; Takeno, H.; Hayasaka, O.; Kotani, T.; et al. Neuropeptide Y Deficiency Induces Anxiety-like Behaviours in Zebrafish (Danio rerio). Sci. Rep. 2020, 10, 5913. [Google Scholar] [CrossRef] [Green Version]
- Pedragosa-Badia, X.; Stichel, J.; Beck-Sickinger, A.G. Neuropeptide Y Receptors: How to Get Subtype Selectivity. Front. Endocrinol. 2013, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Miller, R.J. Is There Really an NPY Y3 Receptor? Regul. Pept. 1998, 75–76, 71–78. [Google Scholar] [CrossRef]
- Starbäck, P.; Wraith, A.; Eriksson, H.; Larhammar, D. Neuropeptide Y Receptor Gene y6: Multiple Deaths or Resurrections? Biochem. Biophys. Res. Commun. 2000, 277, 264–269. [Google Scholar] [CrossRef]
- dos Santos, V.V.; Santos, D.B.; Lach, G.; Rodrigues, A.L.S.; Farina, M.; De Lima, T.C.M.; Prediger, R. Neuropeptide Y (NPY) Prevents Depressive-like Behavior, Spatial Memory Deficits and Oxidative Stress Following Amyloid-β (Aβ1–40) Administration in Mice. Behav. Brain Res. 2013, 244, 107–115. [Google Scholar] [CrossRef]
- Malva, J.O.; Xapelli, S.; Baptista, S.; Valero, J.; Agasse, F.; Ferreira, R.; Silva, A.P. Multifaces of Neuropeptide Y in the Brain—Neuroprotection, Neurogenesis and Neuroinflammation. Neuropeptides 2012, 46, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, S.L.; Tyce, G.M.; Ahlskog, J.; Zinsmeister, A.R.; Nelson, D.K.; Carmichael, S.W. Decreased Levels of [Met]Enkephalin, Neuropeptide Y, Substance P, and Vasoactive Intestinal Peptide in Parkinsonian Adrenal Medulla. Exp. Neurol. 1991, 114, 23–27. [Google Scholar] [CrossRef]
- Martignoni, E.; Blandini, F.; Petraglia, F.; Pacchetti, C.; Bono, G.; Nappi, G. Cerebrospinal Fluid Norepinephrine, 3-Methoxy-4-Hydroxyphenylglycol and Neuropeptide Y Levels in Parkinson’s Disease, Multiple System Atrophy and Dementia of the Alzheimer Type. J. Neural Transm. 1992, 4, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Cannizzaro, C.; Tel, B.C.; Rose, S.; Zeng, B.-Y.; Jenner, P. Increased Neuropeptide Y mRNA Expression in Striatum in Parkinson’s Disease. Mol. Brain Res. 2003, 110, 169–176. [Google Scholar] [CrossRef]
- Goto, S.; Kawarai, T.; Morigaki, R.; Okita, S.; Koizumi, H.; Nagahiro, S.; Munoz, E.L.; Lee, L.V.; Kaji, R. Defects in the Striatal Neuropeptide Y System in X-Linked Dystonia-Parkinsonism. Brain 2013, 136, 1555–1567. [Google Scholar] [CrossRef] [Green Version]
- Svenningsson, P.; Pålhagen, S.; Mathé, A.A. Neuropeptide Y and Calcitonin Gene-Related Peptide in Cerebrospinal Fluid in Parkinson’s Disease with Comorbid Depression versus Patients with Major Depressive Disorder. Front. Psychiatry 2017, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Obuchowicz, E.; Antkiewicz-Michaluk, L.; Romańska, I.; Herman, Z.S. Increased Striatal Neuropeptide Y Immunoreactivity and Its Modulation by Deprenyl, Clonidine and L-dopa in MPTP-treated Mice. J. Neural Transm. 2003, 110, 1375–1391. [Google Scholar] [CrossRef]
- Decressac, M.; Pain, S.; Chabeauti, P.-Y.; Frangeul, L.; Thiriet, N.; Herzog, H.; Vergote, J.; Chalon, S.; Jaber, M.; Gaillard, A. Neuroprotection by Neuropeptide Y in Cell and Animal Models of Parkinson’s Disease. Neurobiol. Aging 2012, 33, 2125–2137. [Google Scholar] [CrossRef]
- Pain, S.; Vergote, J.; Gulhan, Z.; Bodard, S.; Chalon, S.; Gaillard, A. Inflammatory Process in Parkinson Disease: Neuroprotection by Neuropeptide Y. Fundam. Clin. Pharmacol. 2019, 33, 544–548. [Google Scholar] [CrossRef]
- Ferreira, R.; Xapelli, S.; Santos, T.; Silva, A.P.; Cristóvão, A.; Cortes, L.; Malva, J.O. Neuropeptide Y Modulation of Interleukin-1β (IL-1β)-induced Nitric Oxide Production in Microglia. J. Biol. Chem. 2010, 285, 41921–41934. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.Y.; Hong, S.H.; Kim, B.; Lee, D.S.; Yu, K.; Lee, K.-S. Neuropeptide Y Mitigates ER Stress–induced Neuronal Cell Death by Activating the PI3K–XBP1 Pathway. Eur. J. Cell Biol. 2018, 97, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Sendtner, M.; Holtmann, B.; Kolbeck, R.; Thoenen, H.; Barde, Y. Brain-Derived Neurotrophic Factor Prevents the Death of Motoneurons in Newborn Rats after Nerve Section. Nature 1992, 360, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Racagni, G.; Riva, M.A. Shedding Light into the Role of BDNF in the Pharmacotherapy of Parkinson’s Disease. Pharm. J. 2006, 6, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraway, R.; Leeman, S.E. The Isolation of a New Hypotensive Peptide, Neurotensin, from Bovine Hypothalami. J. Biol. Chem. 1973, 248, 6854–6861. [Google Scholar] [CrossRef]
- Russjan, E.; Kaczyńska, K. Beneficial Effects of Neurotensin in Murine Model of Hapten-Induced Asthma. Int. J. Mol. Sci. 2019, 20, 5025. [Google Scholar] [CrossRef] [Green Version]
- Carraway, R.; Leeman, S.E. The Amino Acid Sequence of a Hypothalamic Peptide, Neurotensin. J. Biol. Chem. 1975, 250, 1907–1911. [Google Scholar] [CrossRef]
- St.-Gelais, F.; Jomphe, C.; Trudeau, L.É.. The Role of Neurotensin in Central Nervous System Pathophysiology: What Is the Evidence? J. Psychiatry Neurosci. 2006, 31, 229–245. [Google Scholar]
- Wu, Z.; Martinez-Fong, D.; Trédaniel, J.; Forgez, P. Neurotensin and Its High Affinity Receptor 1 as a Potential Pharmacological Target in Cancer Therapy. Front. Endocrinol. 2013, 3, 184. [Google Scholar] [CrossRef] [Green Version]
- Mazella, J.; Béraud-Dufour, S.; Devader, C.; Massa, F.; Coppola, T. Neurotensin and Its Receptors in the Control of Glucose Homeostasis. Front. Endocrinol. 2012, 3, 143. [Google Scholar] [CrossRef] [Green Version]
- Jomphe, C.; Lemelin, P.-L.; Okano, H.; Kobayashi, K.; Trudeau, L.-E. Bidirectional Regulation of Dopamine D2 and Neurotensin NTS1 Receptors in Dopamine Neurons. Eur. J. Neurosci. 2006, 24, 2789–2800. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.; de Ceballos, M.L.; Rose, S.; Jenner, P.; Marsden, C.D. Alterations in Peptide Levels in Parkinson’s Disease and Incidental Lewy Body Disease. Brain 1996, 119, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.; Jenner, P.; Marsden, C.D.; de Ceballos, M.L. Characterization of Neurotensin-like Immunoreactivity in Human Basal Ganglia: Increased Neurotensin Levels in Substantia Nigra in Parkinson’s Disease. Peptides 1995, 16, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Schimpff, R.-M.; Avard, C.; Fénelon, G.; Lhiaubet, A.-M.; Tennezé, L.; Vidailhet, M.; Rostène, W. Increased Plasma Neurotensin Concentrations in Patients with Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2001, 70, 784–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Richelson, E. Heterogeneity of Melanized Neurons Expressing Neurotensin Receptor Messenger RNA in the Substantia Nigra and the Nucleus Paranigralis of Control and Parkinson’s Disease Brain. Neuroscience 1995, 64, 405–417. [Google Scholar] [CrossRef]
- Uhl, G.R.; Whitehouse, P.J.; Price, D.L.; Tourtelotte, W.W.; Kuhar, M.J. Parkinson’s Disease: Depletion of Substantia Nigra Neurotensin Receptors. Brain Res. 1984, 308, 186–190. [Google Scholar] [CrossRef]
- Fernandez, A.; de Ceballos, M.L.; Jenner, P.; Marsden, C.D. Neurotensin, Substance P, Delta and Mu Opioid Receptors Are Decreased in Basal Ganglia of Parkinson’s Disease Patients. Neuroscience 1994, 61, 73–79. [Google Scholar] [CrossRef]
- Chinaglia, G.; Probst, A.; Palacios, J. Neurotensin Receptors in Parkinson’s Disease and Progressive Supranuclear Palsy: An Autoradiographic Study in Basal Ganglia. Neuroscience 1990, 39, 351–360. [Google Scholar] [CrossRef]
- Rivest, R.; St-Pierre, S.; Jolicoeur, F.B. Structure-Activity Studies of Neurotensin on Muscular Rigidity and Tremors Induced by 6-Hydroxydopamine Lesions in the Posterolateral Hypothalamus of the Rat. Neuropharmacology 1991, 30, 47–52. [Google Scholar] [CrossRef]
- Antonelli, T.; Fuxe, K.; Tomasini, M.C.; Mazzoni, E.; Agnati, L.F.; Tanganelli, S.; Ferraro, L. Neurotensin Receptor Mechanisms and Its Modulation of Glutamate Transmission in the Brain: Relevance for Neurodegenerative Diseases and Their Treatment. Prog. Neurobiol. 2007, 83, 92–109. [Google Scholar] [CrossRef]
- Gilmartin, M.R.; Ferrara, N.C. Pituitary Adenylate Cyclase-Activating Polypeptide in Learning and Memory. Front. Cell. Neurosci. 2021, 15, 663418. [Google Scholar] [CrossRef]
- Miyata, A.; Arimura, A.; Dahl, R.R.; Minamino, N.; Uehara, A.; Jiang, L.; Culler, M.D.; Coy, D.H. Isolation of a Novel 38 Residue-Hypothalamic Polypeptide Which Stimulates Adenylate Cyclase in Pituitary Cells. Biochem. Biophys. Res. Commun. 1989, 164, 567–574. [Google Scholar] [CrossRef]
- Liao, C.; De Molliens, M.P.; Schneebeli, S.T.; Brewer, M.; Song, G.; Chatenet, D.; Braas, K.M.; May, V.; Li, J. Targeting the PAC1 Receptor for Neurological and Metabolic Disorders. Curr. Top. Med. Chem. 2019, 19, 1399–1417. [Google Scholar] [CrossRef] [PubMed]
- Shioda, S.; Takenoya, F.; Wada, N.; Hirabayashi, T.; Seki, T.; Nakamachi, T. Pleiotropic and Retinoprotective Functions of PACAP. Anat. Sci. Int. 2016, 91, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Lamine-Ajili, A.; Fahmy, A.M.; Létourneau, M.; Chatenet, D.; Labonté, P.; Vaudry, D.; Fournier, A. Effect of the Pituitary Adenylate Cyclase-Activating Polypeptide on the Autophagic Activation Observed in in Vitro and in Vivo Models of Parkinson’s Disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, T.; Shibato, J.; Kimura, A.; Yamashita, M.; Takenoya, F.; Shioda, S. Potential Therapeutic Role of Pituitary Adenylate Cyclase-Activating Polypeptide for Dry Eye Disease. Int. J. Mol. Sci. 2022, 23, 664. [Google Scholar] [CrossRef] [PubMed]
- Saklani, P.; Khan, H.; Gupta, S.; Kaur, A.; Singh, T.G. Neuropeptides: Potential Neuroprotective Agents in Ischemic Injury. Life Sci. 2022, 288, 120186. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, J. Pituitary Adenylate Cyclase-Activating Peptide in the Rat Central Nervous System: An Immunohistochemical and in Situ Hybridization Study. J. Comp. Neurol. 2002, 453, 389–417. [Google Scholar] [CrossRef]
- Feher, M.; Gaszner, B.; Tamas, A.; Gil-Martinez, A.L.; Fernandez-Villalba, E.; Herrero, M.T.; Reglodi, D. Alteration of the PAC1 Receptor Expression in the Basal Ganglia of MPTP-Induced Parkinsonian Macaque Monkeys. Neurotox. Res. 2018, 33, 702–715. [Google Scholar] [CrossRef]
- Reglodi, D.; Renaud, J.; Tamas, A.; Tizabi, Y.; Socías, S.B.; Del-Bel, E.; Raisman-Vozari, R. Novel Tactics for Neuroprotection in Parkinson’s Disease: Role of Antibiotics, Polyphenols and Neuropeptides. Prog. Neurobiol. 2017, 155, 120–148. [Google Scholar] [CrossRef]
- Reglodi, D.; Kiss, P.; Lubics, A.; Tamas, A. Review on the Protective Effects of PACAP in Models of Neurodegenerative Diseases In Vitro and In Vivo. Curr. Pharm. Des. 2011, 17, 962–972. [Google Scholar] [CrossRef]
- Reglodi, D.; Lubics, A.; Tamás, A.; Szalontay, L.; Lengvári, I. Pituitary Adenylate Cyclase Activating Polypeptide Protects Dopaminergic Neurons and Improves Behavioral Deficits in a Rat Model of Parkinson’s Disease. Behav. Brain Res. 2004, 151, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Reglődi, D.; Tamás, A.; Lubics, A.; Szalontay, L.; Lengvári, I. Morphological and Functional Effects of PACAP in 6-Hydroxydopamine-Induced Lesion of the Substantia Nigra in Rats. Regul. Pept. 2004, 123, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Deguil, J.; Chavant, F.; Lafay-Chebassier, C.; Pérault-Pochat, M.-C.; Fauconneau, B.; Pain, S. Neuroprotective Effect of PACAP on Translational Control Alteration and Cognitive Decline in MPTP Parkinsonian Mice. Neurotox. Res. 2010, 17, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Qi, C.; Fan, G.-H.; Zhou, H.-Y.; Chen, S.-D. PACAP Protects Neuronal Differentiated PC12 Cells against the Neurotoxicity Induced by a Mitochondrial Complex I Inhibitor, Rotenone. FEBS Lett. 2005, 579, 4005–4011. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Tamas, A.; Reglodi, D.; Tizabi, Y. PACAP Protects Against Salsolinol-Induced Toxicity in Dopaminergic SH-SY5Y Cells: Implication for Parkinson’s Disease. J. Mol. Neurosci. 2013, 50, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Shivers, K.-Y.; Nikolopoulou, A.; Machlovi, S.I.; Vallabhajosula, S.; Figueiredo-Pereira, M.E. PACAP27 Prevents Parkinson-like Neuronal Loss and Motor Deficits but Not Microglia Activation Induced by Prostaglandin J2. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2014, 1842, 1707–1719. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Tamás, A.; Reglodi, D.; Tizabi, Y. PACAP Protects Against Inflammatory-Mediated Toxicity in Dopaminergic SH-SY5Y Cells: Implication for Parkinson’s Disease. Neurotox. Res. 2014, 26, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Pan, J.; Tan, Y.-Y.; Sun, X.-K.; Zhang, Y.-F.; Zhou, H.-Y.; Ren, R.-J.; Wang, X.-J.; Chen, S.-D. Neuroprotective Effects of PACAP27 in Mice Model of Parkinson’s Disease Involved in the Modulation of KATP Subunits and D2 Receptors in the Striatum. Neuropeptides 2008, 42, 267–276. [Google Scholar] [CrossRef]
- Yang, S.; Yang, J.; Yang, Z.; Chen, P.; Fraser, A.; Zhang, W.; Pang, H.; Gao, X.; Wilson, B.; Hong, J.-S.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) 38 and PACAP4–6 Are Neuroprotective through Inhibition of NADPH Oxidase: Potent Regulators of Microglia-Mediated Oxidative Stress. J. Pharmacol. Exp. Ther. 2006, 319, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Said, S.I.; Mutt, V. Polypeptide with Broad Biological Activity: Isolation from Small Intestine. Science 1970, 169, 1217–1218. [Google Scholar] [CrossRef]
- Korkmaz, O.T.; Tunçel, N. Advantages of Vasoactive Intestinal Peptide for the Future Treatment of Parkinson’s Disease. Curr. Pharm. Des. 2019, 24, 4693–4701. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Ganea, D. Neuroprotective Effect of Vasoactive Intestinal Peptide (VIP) In a Mouse Model of Parkinson’s Disease by Blocking Microglial Activation. FASEB J. 2003, 17, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broome, S.T.; Musumeci, G.; Castorina, A. PACAP and VIP Mitigate Rotenone-Induced Inflammation in BV-2 Microglial Cells. J. Mol. Neurosci. 2022, 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.S.; Shimizu, H.; Satoh, T.; Okada, S.; Adachi, S.; Inoue, K.; Eguchi, H.; Yamamoto, M.; Imaki, T.; Hashimoto, K.; et al. Identification of Nesfatin-1 as a Satiety Molecule in the Hypothalamus. Nature 2006, 443, 709–712. [Google Scholar] [CrossRef]
- Rupp, S.K.; Wölk, E.; Stengel, A. Nesfatin-1 Receptor: Distribution, Signaling and Increasing Evidence for a G Protein-Coupled Receptor—A Systematic Review. Front. Endocrinol. 2021, 12, 740174. [Google Scholar] [CrossRef]
- Goebel-Stengel, M.; Wang, L. Central and Peripheral Expression and Distribution of NUCB2/nesfatin-1. Curr. Pharm. Des. 2013, 19, 6935–6940. [Google Scholar] [CrossRef]
- Shimizu, H.; Mori, M. Nesfatin-1: Its Role in the Diagnosis and Treatment of Obesity and Some Psychiatric Disorders. Calcium-Bind. Proteins RAGE 2013, 963, 327–338. [Google Scholar] [CrossRef]
- Chen, H.; Li, X.; Ma, H.; Zheng, W.; Shen, X. Reduction in Nesfatin-1 Levels in the Cerebrospinal Fluid and Increased Nigrostriatal Degeneration Following Ventricular Administration of Anti-nesfatin-1 Antibody in Mice. Front. Neurosci. 2021, 15, 621173. [Google Scholar] [CrossRef]
- Price, T.O.; Samson, W.K.; Niehoff, M.L.; Banks, W.A. Permeability of the Blood–Brain Barrier to a Novel Satiety Molecule Nesfatin-1. Peptides 2007, 28, 2372–2381. [Google Scholar] [CrossRef]
- Emir, G.K.; Ünal, Y.; Yılmaz, N.; Tosun, K.; Kutlu, G. The Association of Low Levels of Nesfatin-1 and Glucagon-like Peptide-1 with Oxidative Stress in Parkinson’s Disease. Neurol. Sci. 2019, 40, 2529–2535. [Google Scholar] [CrossRef]
- Tang, C.-H.; Fu, X.-J.; Xu, X.-L.; Wei, X.-J.; Pan, H.-S. The Anti-inflammatory and Anti-apoptotic Effects of Nesfatin-1 in the Traumatic Rat Brain. Peptides 2012, 36, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Özsavcí, D.; Erşahin, M.; Şener, A.; Özakpinar, Ö.B.; Toklu, H.Z.; Akakín, D.; Şener, G.; Yeğen, B. The Novel Function of Nesfatin-1 as an Anti-inflammatory and Antiapoptotic Peptide in Subarachnoid Hemorrhage–Induced Oxidative Brain Damage in Rats. Neurosurgery 2011, 68, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Xu, H.; Shen, X.; Jiang, H. Nesfatin-1 Antagonized Rotenone-Induced Neurotoxicity in MES23.5 Dopaminergic Cells. Peptides 2015, 69, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Erfani, S.; Moghimi, A.; Aboutaleb, N.; Khaksari, M. Protective Effects of Nesfatin-1 Peptide on Cerebral Ischemia Reperfusion Injury via Inhibition of Neuronal Cell Death and Enhancement of Antioxidant Defenses. Metab. Brain Dis. 2019, 34, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-L.; Song, N.; Du, X.-X.; Li, Y.; Xie, J.-X.; Jiang, H. Nesfatin-1 Protects Dopaminergic Neurons against MPP+/MPTP-Induced Neurotoxicity through the C-Raf–ERK1/2-Dependent Anti-Apoptotic Pathway. Sci. Rep. 2017, 7, 40961. [Google Scholar] [CrossRef] [Green Version]
- Brazeau, P.; Vale, W.; Burgus, R.; Guillemin, R. Isolation of Somatostatin (a Somatotropin Release Inhibiting Factor) of Ovine Hypothalamic Origin. Can. J. Biochem. 1974, 52, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.C. Somatostatin and Its Receptor Family. Front. Neuroendocr. 1999, 20, 157–198. [Google Scholar] [CrossRef]
- Kumar, U.; Grant, M. Somatostatin and Somatostatin Receptors. In Results and Problems in Cell Differentiation; Rehfeld, J., Bundgaard, J., Eds.; Springer: Berlin, Germany, 2010; Volume 50, pp. 137–184. [Google Scholar] [CrossRef]
- O’Toole, T.J.; Sharma, S. Physiology, Somatostatin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Weckbecker, G.; Lewis, I.; Albert, R.; Schmid, H.A.; Hoyer, D.; Bruns, C. Opportunities in Somatostatin Research: Biological, Chemical and Therapeutic Aspects. Nat. Rev. Drug Discov. 2003, 2, 999–1017. [Google Scholar] [CrossRef]
- Reubi, J.C. Somatostatin Receptors in the Gastrointestinal Tract in Health and Disease. Yale J. Biol. Med. 1992, 65, 493–536. [Google Scholar]
- Reichlin, S. Somatostatin (Second of Two Parts). N. Engl. J. Med. 1983, 309, 1556–1563. [Google Scholar] [CrossRef]
- Reichlin, S. Somatostatin (First of Two Parts). N. Engl. J. Med. 1983, 309, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Rachamalla, M.; Najda, A.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Chigurupati, S.; Vargas-De-La-Cruz, C.; et al. Applications of Adductomics in Chemically Induced Adverse Outcomes and Major Emphasis on DNA Adductomics: A Pathbreaking Tool in Biomedical Research. Int. J. Mol. Sci. 2021, 22, 10141. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Makeen, H.A.; Albratty, M.; Alhazmi, H.A.; Meraya, A.M.; Bungau, S. Demystifying the Neuroprotective Role of Neuropeptides in Parkinson’s Disease: A Newfangled and Eloquent Therapeutic Perspective. Int. J. Mol. Sci. 2022, 23, 4565. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094565
Behl T, Madaan P, Sehgal A, Singh S, Makeen HA, Albratty M, Alhazmi HA, Meraya AM, Bungau S. Demystifying the Neuroprotective Role of Neuropeptides in Parkinson’s Disease: A Newfangled and Eloquent Therapeutic Perspective. International Journal of Molecular Sciences. 2022; 23(9):4565. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094565
Chicago/Turabian StyleBehl, Tapan, Piyush Madaan, Aayush Sehgal, Sukhbir Singh, Hafiz A. Makeen, Mohammed Albratty, Hassan A. Alhazmi, Abdulkarim M. Meraya, and Simona Bungau. 2022. "Demystifying the Neuroprotective Role of Neuropeptides in Parkinson’s Disease: A Newfangled and Eloquent Therapeutic Perspective" International Journal of Molecular Sciences 23, no. 9: 4565. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094565