
Role of Glycogen Synthase Kinase-3 in Interferon-γ-Mediated Immune Hepatitis

 ,
,
Abstract
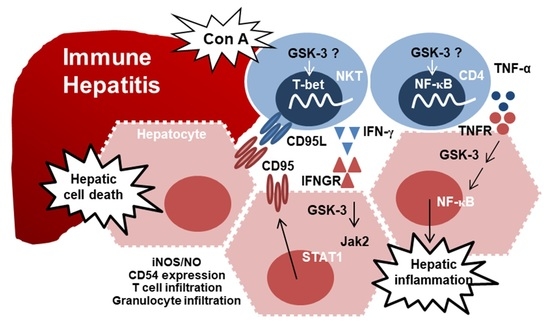
:
1. Multiple Roles of Glycogen Synthase Kinase-3 (GSK-3) in Human Diseases
2. Regulation of GSK-3 in Facilitating Proapoptosis and Proinflammation
3. Targeting GSK-3 as a Protective Strategy against Hepatic Injury
4. Generation of IFN-γ and Its Multiple Proinflammatory Roles
5. IFN-γ Signaling and Its Regulation
6. GSK-3 Is Involved in IFN-γ Signaling Pathways
7. Immune Hepatitis
8. GSK-3 in IFN-γ-Mediated Hepatic Immune Hepatitis and Its Therapeutic Efficacy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ConA | concanavalin A |
| CREB | cAMP-response element-binding protein |
| D-GalN | D-galactosamine |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| GSK | glycogen synthase kinase |
| IFN | interferon |
| IFNGR | IFN-γ receptor |
| IL | interleukin |
| iNOS | inducible NO synthase |
| IRF | interferon regulatory factor |
| IRI | ischemia/reperfusion injury |
| LiCl | lithium chloride |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| NF-κB | nuclear factor κB |
| NK | natural killer |
| NO | nitric oxide |
| PI3K | phosphatidylinositol 3-kinase |
| PK | protein kinase |
| PMNs | polymorphonuclear granulocytes |
| PP | protein phosphatase |
| Pyk | proline-rich tyrosine kinase |
| RANTES | regulated on activation, normal T-cell expressed and secreted |
| SHP | SH2-containing phosphatase |
| SOCS | suppressor of cytokine signaling |
| STAT | signal transducer and activator of transcription |
| T-bet | T-box transcription factor Tbx21 |
| TDZD-8 | 4-Benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
References
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijur, G.N.; Jope, R.S. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport 2003, 14, 2415–2419. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Kockeritz, L.; Doble, B.; Patel, S.; Woodgett, J.R. Glycogen synthase kinase-3—An overview of an over-achieving protein kinase. Curr. Drug Targets 2006, 7, 1377–1388. [Google Scholar] [CrossRef]
- Jope, R.S.; Johnson, G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [Green Version]
- Emma, M.R.; Augello, G.; Cusimano, A.; Azzolina, A.; Montalto, G.; McCubrey, J.A.; Cervello, M. GSK-3 in liver diseases: Friend or foe? Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118743. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Hetman, M.; Hsuan, S.L.; Habas, A.; Higgins, M.J.; Xia, Z. ERK1/2 antagonizes glycogen synthase kinase-3beta-induced apoptosis in cortical neurons. J. Biol. Chem. 2002, 277, 49577–49584. [Google Scholar] [CrossRef] [Green Version]
- Pap, M.; Cooper, G.M. Role of translation initiation factor 2B in control of cell survival by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta signaling pathway. Mol. Cell. Biol. 2002, 22, 578–586. [Google Scholar] [CrossRef] [Green Version]
- Ivaska, J.; Nissinen, L.; Immonen, N.; Eriksson, J.E.; Kahari, V.M.; Heino, J. Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol. Cell. Biol. 2002, 22, 1352–1359. [Google Scholar] [CrossRef] [Green Version]
- Hartigan, J.A.; Xiong, W.C.; Johnson, G.V. Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem. Biophys. Res. Commun. 2001, 284, 485–489. [Google Scholar] [CrossRef]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 2004, 377, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Bijur, G.N.; De Sarno, P.; Jope, R.S. Glycogen synthase kinase-3beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J. Biol. Chem. 2000, 275, 7583–7590. [Google Scholar] [CrossRef] [Green Version]
- Somervaille, T.C.; Linch, D.C.; Khwaja, A. Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood 2001, 98, 1374–1381. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; De Sarno, P.; Jope, R.S. Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 2002, 277, 44701–44708. [Google Scholar] [CrossRef] [Green Version]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Chen, C.L.; Chiang, C.W.; Jan, M.S.; Huang, W.C.; Lin, Y.S. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J. Cell Sci. 2007, 120, 2935–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.C.; Lin, Y.S.; Chen, C.L.; Wang, C.Y.; Chiu, W.H.; Lin, C.F. Glycogen synthase kinase-3beta mediates endoplasmic reticulum stress-induced lysosomal apoptosis in leukemia. J. Pharmacol. Exp. Ther. 2009, 329, 524–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugo, L.; Collin, M.; Allen, D.A.; Patel, N.S.; Bauer, I.; Mervaala, E.M.; Louhelainen, M.; Foster, S.J.; Yaqoob, M.M.; Thiemermann, C. GSK-3beta inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit. Care Med. 2005, 33, 1903–1912. [Google Scholar] [CrossRef]
- De Sarno, P.; Axtell, R.C.; Raman, C.; Roth, K.A.; Alessi, D.R.; Jope, R.S. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2008, 181, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Whittle, B.J.; Varga, C.; Posa, A.; Molnar, A.; Collin, M.; Thiemermann, C. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3beta. Br. J. Pharmacol. 2006, 147, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Takada, Y.; Fang, X.; Jamaluddin, M.S.; Boyd, D.D.; Aggarwal, B.B. Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J. Biol. Chem. 2004, 279, 39541–39554. [Google Scholar] [CrossRef] [Green Version]
- Cuzzocrea, S.; Mazzon, E.; Di Paola, R.; Muia, C.; Crisafulli, C.; Dugo, L.; Collin, M.; Britti, D.; Caputi, A.P.; Thiemermann, C. Glycogen synthase kinase-3beta inhibition attenuates the degree of arthritis caused by type II collagen in the mouse. Clin. Immunol. 2006, 120, 57–67. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Woodgett, J.R.; Ohashi, P.S. GSK3: An in-Toll-erant protein kinase? Nat. Immunol. 2005, 6, 751–752. [Google Scholar] [CrossRef]
- Huang, W.C.; Lin, Y.S.; Wang, C.Y.; Tsai, C.C.; Tseng, H.C.; Chen, C.L.; Lu, P.J.; Chen, P.S.; Qian, L.; Hong, J.S.; et al. Glycogen synthase kinase-3 negatively regulates anti-inflammatory interleukin-10 for lipopolysaccharide-induced iNOS/NO biosynthesis and RANTES production in microglial cells. Immunology 2009, 128, e275–e286. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, W.C.; Wang, C.Y.; Tsai, C.C.; Chen, C.L.; Chang, Y.T.; Kai, J.I.; Lin, C.F. Inhibiting glycogen synthase kinase-3 reduces endotoxaemic acute renal failure by down-regulating inflammation and renal cell apoptosis. Br. J. Pharmacol. 2009, 157, 1004–1013. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.L.; Wang, C.Y.; Huang, W.C.; Tsai, C.C.; Chen, C.L.; Shen, C.F.; Chi, C.Y.; Lin, C.F. Staphylococcus aureus induces microglial inflammation via a glycogen synthase kinase 3beta-regulated pathway. Infect. Immun. 2009, 77, 4002–4008. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.F.; Tsai, C.C.; Huang, W.C.; Wang, C.Y.; Tseng, H.C.; Wang, Y.; Kai, J.I.; Wang, S.W.; Cheng, Y.L. IFN-gamma synergizes with LPS to induce nitric oxide biosynthesis through glycogen synthase kinase-3-inhibited IL-10. J. Cell. Biochem. 2008, 105, 746–755. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Di Paola, R.; Mazzon, E.; Crisafulli, C.; Genovese, T.; Muia, C.; Abdelrahman, M.; Esposito, E.; Thiemermann, C. Glycogen synthase kinase 3beta inhibition reduces the development of nonseptic shock induced by zymosan in mice. Shock 2007, 27, 97–107. [Google Scholar] [CrossRef]
- Ren, F.; Duan, Z.; Cheng, Q.; Shen, X.; Gao, F.; Bai, L.; Liu, J.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Zhai, Y. Inhibition of glycogen synthase kinase 3 beta ameliorates liver ischemia reperfusion injury by way of an interleukin-10-mediated immune regulatory mechanism. Hepatology 2011, 54, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Joe, Y.; Kong, J.S.; Jeong, S.O.; Cho, G.J.; Ryter, S.W.; Chung, H.T. Carbon monoxide protects against hepatic ischemia/reperfusion injury via ROS-dependent Akt signaling and inhibition of glycogen synthase kinase 3beta. Oxid. Med. Cell. Longev. 2013, 2013, 306421. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tassiulas, I.; Park-Min, K.H.; Reid, A.C.; Gil-Henn, H.; Schlessinger, J.; Baron, R.; Zhang, J.J.; Ivashkiv, L.B. ‘Tuning’ of type I interferon-induced Jak-STAT1 signaling by calcium-dependent kinases in macrophages. Nat. Immunol. 2008, 9, 186–193. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, L.; Jin, P.; Li, N.; Meng, Y.; Wang, C.; Xu, M.; Zhang, Y.; Bian, J.; Deng, X. Methane alleviates carbon tetrachloride induced liver injury in mice: Anti-inflammatory action demonstrated by increased PI3K/Akt/GSK-3beta-mediated IL-10 expression. J. Mol. Histol. 2017, 48, 301–310. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yahata, H.; Tanji, H.; Sakaguchi, T.; Ito, H.; Dohi, K. Allograft transduction of IL-10 prolongs survival following orthotopic liver transplantation. Gene Ther. 1999, 6, 816–822. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.S.; Jeong, I.S.; Han, J.H.; Cheon, S.H.; Kim, S.W. IL-10-secreting human MSCs generated by TALEN gene editing ameliorate liver fibrosis through enhanced anti-fibrotic activity. Biomater. Sci. 2019, 7, 1078–1087. [Google Scholar] [CrossRef]
- Louis, H.; Le Moine, O.; Peny, M.O.; Quertinmont, E.; Fokan, D.; Goldman, M.; Deviere, J. Production and role of interleukin-10 in concanavalin A-induced hepatitis in mice. Hepatology 1997, 25, 1382–1389. [Google Scholar] [CrossRef]
- Ren, F.; Zhou, L.; Zhang, X.; Wen, T.; Shi, H.; Xie, B.; Li, Z.; Chen, D.; Wang, Z.; Duan, Z. Endoplasmic reticulum stress-activated glycogen synthase kinase 3beta aggravates liver inflammation and hepatotoxicity in mice with acute liver failure. Inflammation 2015, 38, 1151–1165. [Google Scholar] [CrossRef]
- Chen, L.; Ren, F.; Zhang, H.; Wen, T.; Piao, Z.; Zhou, L.; Zheng, S.; Zhang, J.; Chen, Y.; Han, Y.; et al. Inhibition of glycogen synthase kinase 3beta ameliorates D-GalN/LPS-induced liver injury by reducing endoplasmic reticulum stress-triggered apoptosis. PLoS ONE 2012, 7, e45202. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.H.; Gong, J.P.; Li, J.Z.; He, K.; Li, P.Z.; Jiang, X.W. Glycogen synthase kinase 3 inhibitor attenuates endotoxin-induced liver injury. J. Surg. Res. 2013, 184, 1035–1044. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, W.; Fang, H.; Yang, Y.; Li, X.; He, J.; Jiang, X.; Wang, W.; Liu, S.; Hu, J.; et al. GSK-3beta inhibition attenuates CLP-induced liver injury by reducing inflammation and hepatic cell apoptosis. Mediat. Inflamm. 2014, 2014, 629507. [Google Scholar] [CrossRef] [Green Version]
- Jellestad, L.; Fink, T.; Pradarutti, S.; Kubulus, D.; Wolf, B.; Bauer, I.; Thiemermann, C.; Rensing, H. Inhibition of glycogen synthase kinase (GSK)-3-beta improves liver microcirculation and hepatocellular function after hemorrhagic shock. Eur. J. Pharmacol. 2014, 724, 175–184. [Google Scholar] [CrossRef]
- Rocha, J.; Figueira, M.E.; Barateiro, A.; Fernandes, A.; Brites, D.; Pinto, R.; Freitas, M.; Fernandes, E.; Mota-Filipe, H.; Sepodes, B. Inhibition of glycogen synthase kinase-3beta attenuates organ injury and dysfunction associated with liver ischemia-reperfusion and thermal injury in the rat. Shock 2015, 43, 369–378. [Google Scholar] [CrossRef]
- Ren, F.; Zhang, L.; Zhang, X.; Shi, H.; Wen, T.; Bai, L.; Zheng, S.; Chen, Y.; Chen, D.; Li, L.; et al. Inhibition of glycogen synthase kinase 3beta promotes autophagy to protect mice from acute liver failure mediated by peroxisome proliferator-activated receptor alpha. Cell Death Dis. 2016, 7, e2151. [Google Scholar] [CrossRef]
- Wei, L.; Ren, F.; Zhang, X.; Wen, T.; Shi, H.; Zheng, S.; Zhang, J.; Chen, Y.; Han, Y.; Duan, Z. Oxidative stress promotes D-GalN/LPS-induced acute hepatotoxicity by increasing glycogen synthase kinase 3 beta activity. Inflamm. Res. 2014, 63, 485–494. [Google Scholar] [CrossRef]
- Wang, J.; Deng, M.; Wu, H.; Wang, M.; Gong, J.; Bai, H.; Wu, Y.; Pan, J.; Chen, Y.; Li, S. Suberoylanilide hydroxamic acid alleviates orthotopic liver transplantationinduced hepatic ischemiareperfusion injury by regulating the AKT/GSK3beta/NFkappaB and AKT/mTOR pathways in rat Kupffer cells. Int. J. Mol. Med. 2020, 45, 1875–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhusaini, A.; Fadda, L.; Hasan, I.H.; Zakaria, E.; Alenazi, A.M.; Mahmoud, A.M. Curcumin ameliorates lead-induced hepatotoxicity by suppressing oxidative stress and inflammation, and modulating Akt/GSK-3 beta signaling pathway. Biomolecules 2019, 9, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Emam, R.A.; Ali, M.F. Effect of l-carnitine supplementation on lead acetate-induced liver cell apoptosis and inflammation: Role of caspase-3 and glycogen synthase kinase-3beta enzymes. Life Sci. 2022, 291, 120277. [Google Scholar] [CrossRef]
- Jiang, Y.; Bao, H.; Ge, Y.; Tang, W.; Cheng, D.; Luo, K.; Gong, G.; Gong, R. Therapeutic targeting of GSK3beta enhances the Nrf2 antioxidant response and confers hepatic cytoprotection in hepatitis C. Gut 2015, 64, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Jiang, Y.F.; Ponnusamy, M.; Diallo, M. Role of Nrf2 in chronic liver disease. World J. Gastroenterol. 2014, 20, 13079–13087. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Akazawa, Y.; Cazanave, S.C.; Bronk, S.F.; Elmi, N.A.; Werneburg, N.W.; Billadeau, D.D.; Gores, G.J. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J. Hepatol. 2011, 54, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Herrero, C.; Hu, X.; Li, W.P.; Samuels, S.; Sharif, M.N.; Kotenko, S.; Ivashkiv, L.B. Reprogramming of IL-10 activity and signaling by IFN-gamma. J. Immunol. 2003, 171, 5034–5041. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Boothby, M. The calculus of integrating differentiation: Timing control of T-bet. Immunity 2009, 30, 666–668. [Google Scholar] [CrossRef] [Green Version]
- Siebler, J.; Wirtz, S.; Klein, S.; Protschka, M.; Blessing, M.; Galle, P.R.; Neurath, M.F. A key pathogenic role for the STAT1/T-bet signaling pathway in T-cell-mediated liver inflammation. Hepatology 2003, 38, 1573–1580. [Google Scholar]
- Szabo, S.J.; Sullivan, B.M.; Stemmann, C.; Satoskar, A.R.; Sleckman, B.P.; Glimcher, L.H. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 2002, 295, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.J.; Weinmann, A.S.; Matsuda, J.L.; Salomon, R.; Farnham, P.J.; Biron, C.A.; Gapin, L.; Glimcher, L.H. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity 2004, 20, 477–494. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.G.; Mariani, L.; Radbruch, A.; Hofer, T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity 2009, 30, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shier, P.; Hofstra, C.L.; Ma, X.J.; Wu, Y.; Ngo, K.; Fung-Leung, W.P. Tbt-1, a new T-box transcription factor induced in activated Th1 and CD8+ T cells. Immunogenetics 2000, 51, 771–778. [Google Scholar] [CrossRef]
- Wang, J.; Fathman, J.W.; Lugo-Villarino, G.; Scimone, L.; von Andrian, U.; Dorfman, D.M.; Glimcher, L.H. Transcription factor T-bet regulates inflammatory arthritis through its function in dendritic cells. J. Clin. Investig. 2006, 116, 414–421. [Google Scholar] [CrossRef]
- Peng, S.L.; Szabo, S.J.; Glimcher, L.H. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc. Natl. Acad. Sci. USA 2002, 99, 5545–5550. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, J.L.; George, T.C.; Hagman, J.; Gapin, L. Temporal dissection of T-bet functions. J. Immunol. 2007, 178, 3457–3465. [Google Scholar] [CrossRef] [Green Version]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar]
- Jaruga, B.; Hong, F.; Kim, W.H.; Gao, B. IFN-gamma/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: A critical role of IRF-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1044–G1052. [Google Scholar] [CrossRef]
- Hwang, E.S.; Hong, J.H.; Glimcher, L.H. IL-2 production in developing Th1 cells is regulated by heterodimerization of RelA and T-bet and requires T-bet serine residue 508. J. Exp. Med. 2005, 202, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Rose, J.L.; Hoyt, D.G. p38 Mitogen-activated protein kinase mediates synergistic induction of inducible nitric-oxide synthase by lipopolysaccharide and interferon-gamma through signal transducer and activator of transcription 1 Ser727 phosphorylation in murine aortic endothelial cells. Mol. Pharmacol. 2004, 66, 302–311. [Google Scholar] [PubMed]
- Held, T.K.; Weihua, X.; Yuan, L.; Kalvakolanu, D.V.; Cross, A.S. Gamma interferon augments macrophage activation by lipopolysaccharide by two distinct mechanisms, at the signal transduction level and via an autocrine mechanism involving tumor necrosis factor alpha and interleukin-1. Infect. Immun. 1999, 67, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.D.; Riches, D.W. IFN-gamma + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38(mapk) in a mouse macrophage cell line. Am. J. Physiol. Cell Physiol. 2001, 280, C441–C450. [Google Scholar] [CrossRef]
- Koide, N.; Mu, M.M.; Hassan, F.; Islam, S.; Tumurkhuu, G.; Dagvadorj, J.; Naiki, Y.; Mori, I.; Yoshida, T.; Yokochi, T. Lipopolysaccharide enhances interferon-gamma-induced nitric oxide (NO) production in murine vascular endothelial cells via augmentation of interferon regulatory factor-1 activation. J. Endotoxin Res. 2007, 13, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 1997, 15, 749–795. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Jope, R.S. Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 2008, 283, 21934–21944. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, K.; Garotta, G.; Ozmen, L.; Ziemiecki, A.; Wilks, A.F.; Harpur, A.G.; Larner, A.C.; Finbloom, D.S. Interferon-gamma induces tyrosine phosphorylation of interferon-gamma receptor and regulated association of protein tyrosine kinases, Jak1 and Jak2, with its receptor. J. Biol. Chem. 1994, 269, 14333–14336. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Wormald, S.; Hilton, D.J. Inhibitors of cytokine signal transduction. J. Biol. Chem. 2004, 279, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Shiono, S.; Joo, A.; Yoshimura, A. Control mechanism of JAK/STAT signal transduction pathway. FEBS Lett. 2003, 534, 190–196. [Google Scholar] [CrossRef]
- Krebs, D.L.; Hilton, D.J. SOCS: Physiological suppressors of cytokine signaling. J. Cell Sci. 2000, 113 Pt 16, 2813–2819. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Sasaki, A.; Yoshimura, A. Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol. 2000, 18, 143–164. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Yu, D.H.; Feng, G.S. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol. Cell. Biol. 1999, 19, 2416–2424. [Google Scholar] [CrossRef] [Green Version]
- Bennett, A.M.; Tang, T.L.; Sugimoto, S.; Walsh, C.T.; Neel, B.G. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc. Natl. Acad. Sci. USA 1994, 91, 7335–7339. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Paik, P.K.; Chen, J.; Yarilina, A.; Kockeritz, L.; Lu, T.T.; Woodgett, J.R.; Ivashkiv, L.B. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity 2006, 24, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Coant, N.; Simon-Rudler, M.; Gustot, T.; Fasseu, M.; Gandoura, S.; Ragot, K.; Abdel-Razek, W.; Thabut, D.; Letteron, P.; Ogier-Denis, E.; et al. Glycogen synthase kinase 3 involvement in the excessive proinflammatory response to LPS in patients with decompensated cirrhosis. J. Hepatol. 2011, 55, 784–793. [Google Scholar] [CrossRef]
- Hu, X.; Chen, J.; Wang, L.; Ivashkiv, L.B. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J. Leukoc. Biol. 2007, 82, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Brown, J.; Martin, M. Glycogen synthase kinase 3: A point of convergence for the host inflammatory response. Cytokine 2011, 53, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.C.; Kai, J.I.; Huang, W.C.; Wang, C.Y.; Wang, Y.; Chen, C.L.; Fang, Y.T.; Lin, Y.S.; Anderson, R.; Chen, S.H.; et al. Glycogen synthase kinase-3beta facilitates IFN-gamma-induced STAT1 activation by regulating Src homology-2 domain-containing phosphatase 2. J. Immunol. 2009, 183, 856–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayas, C.L.; Ariaens, A.; Ponsioen, B.; Moolenaar, W.H. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol. Biol. Cell 2006, 17, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, A.; Tanaka, N.; Mitani, Y.; Miyazaki, T.; Fujii, H.; Sato, M.; Kovarik, P.; Decker, T.; Schlessinger, J.; Taniguchi, T. Protein tyrosine kinase Pyk2 mediates the Jak-dependent activation of MAPK and Stat1 in IFN-gamma, but not IFN-alpha, signaling. EMBO J. 1999, 18, 2480–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, D.J.; Levy, D.E.; Johnstone, R.W.; Clarke, C.J. IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008, 19, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Patel, T. Apoptosis in hepatic pathophysiology. Clin. Liver Dis. 2000, 4, 295–317. [Google Scholar] [CrossRef]
- Dong, Z.; Wei, H.; Sun, R.; Tian, Z. The roles of innate immune cells in liver injury and regeneration. Cell. Mol. Immunol. 2007, 4, 241–252. [Google Scholar]
- Zheng, Z.Y.; Weng, S.Y.; Yu, Y. Signal molecule-mediated hepatic cell communication during liver regeneration. World J. Gastroenterol. WJG 2009, 15, 5776–5783. [Google Scholar] [CrossRef]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef]
- Sass, G.; Heinlein, S.; Agli, A.; Bang, R.; Schumann, J.; Tiegs, G. Cytokine expression in three mouse models of experimental hepatitis. Cytokine 2002, 19, 115–120. [Google Scholar] [CrossRef]
- Takeda, K.; Hayakawa, Y.; Van Kaer, L.; Matsuda, H.; Yagita, H.; Okumura, K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc. Natl. Acad. Sci. USA 2000, 97, 5498–5503. [Google Scholar] [CrossRef] [Green Version]
- Kusters, S.; Gantner, F.; Kunstle, G.; Tiegs, G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 1996, 111, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Mizuhara, H.; O’Neill, E.; Seki, N.; Ogawa, T.; Kusunoki, C.; Otsuka, K.; Satoh, S.; Niwa, M.; Senoh, H.; Fujiwara, H. T cell activation-associated hepatic injury: Mediation by tumor necrosis factors and protection by interleukin 6. J. Exp. Med. 1994, 179, 1529–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatada, S.; Ohta, T.; Shiratsuchi, Y.; Hatano, M.; Kobayashi, Y. A novel accessory role of neutrophils in concanavalin A-induced hepatitis. Cell. Immunol. 2005, 233, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Harada, M.; Kawano, T.; Yamashita, M.; Shibata, Y.; Gejyo, F.; Nakayama, T.; Taniguchi, M. Augmentation of Valpha14 NKT cell-mediated cytotoxicity by interleukin 4 in an autocrine mechanism resulting in the development of concanavalin A-induced hepatitis. J. Exp. Med. 2000, 191, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Toyabe, S.; Seki, S.; Iiai, T.; Takeda, K.; Shirai, K.; Watanabe, H.; Hiraide, H.; Uchiyama, M.; Abo, T. Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. J. Immunol. 1997, 159, 1537–1542. [Google Scholar] [PubMed]
- Tsai, C.C.; Huang, W.C.; Chen, C.L.; Hsieh, C.Y.; Lin, Y.S.; Chen, S.H.; Yang, K.C.; Lin, C.F. Glycogen synthase kinase-3 facilitates con a-induced IFN-gamma—Mediated immune hepatic injury. J. Immunol. 2011, 187, 3867–3877. [Google Scholar] [CrossRef] [Green Version]
- Carambia, A.; Herkel, J. CD4 T cells in hepatic immune tolerance. J. Autoimmun. 2010, 34, 23–28. [Google Scholar] [CrossRef]
- Bonder, C.S.; Ajuebor, M.N.; Zbytnuik, L.D.; Kubes, P.; Swain, M.G. Essential role for neutrophil recruitment to the liver in concanavalin A-induced hepatitis. J. Immunol. 2004, 172, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Hatano, M.; Sasaki, S.; Ohata, S.; Shiratsuchi, Y.; Yamazaki, T.; Nagata, K.; Kobayashi, Y. Effects of Kupffer cell-depletion on concanavalin A-induced hepatitis. Cell. Immunol. 2008, 251, 25–30. [Google Scholar] [CrossRef]
- Tiegs, G.; Gantner, F. Immunotoxicology of T cell-dependent experimental liver injury. Exp. Toxicol. Pathol. 1996, 48, 471–476. [Google Scholar] [CrossRef]
- Tagawa, Y.; Kakuta, S.; Iwakura, Y. Involvement of Fas/Fas ligand system-mediated apoptosis in the development of concanavalin A-induced hepatitis. Eur. J. Immunol. 1998, 28, 4105–4113. [Google Scholar] [CrossRef]
- Gantner, F.; Leist, M.; Lohse, A.W.; Germann, P.G.; Tiegs, G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: The role of tumor necrosis factor. Hepatology 1995, 21, 190–198. [Google Scholar] [PubMed]
- Tagawa, Y.; Sekikawa, K.; Iwakura, Y. Suppression of concanavalin A-induced hepatitis in IFN-gamma(-/-) mice, but not in TNF-alpha(-/-) mice: Role for IFN-gamma in activating apoptosis of hepatocytes. J. Immunol. 1997, 159, 1418–1428. [Google Scholar] [PubMed]
- Wu, Z.; Han, M.; Chen, T.; Yan, W.; Ning, Q. Acute liver failure: Mechanisms of immune-mediated liver injury. Liver Int. 2010, 30, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Mita, A.; Hashikura, Y.; Tagawa, Y.; Nakayama, J.; Kawakubo, M.; Miyagawa, S. Expression of Fas ligand by hepatic macrophages in patients with fulminant hepatic failure. Am. J. Gastroenterol. 2005, 100, 2551–2559. [Google Scholar] [CrossRef]
- Kai, J.I.; Huang, W.C.; Tsai, C.C.; Chang, W.T.; Chen, C.L.; Lin, C.F. Glycogen synthase kinase-3beta indirectly facilitates interferon-gamma-induced nuclear factor-kappaB activation and nitric oxide biosynthesis. J. Cell. Biochem. 2010, 111, 1522–1530. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H. Glycogen synthase kinase 3: An emerging therapeutic target. Trends Mol. Med. 2002, 8, 126–132. [Google Scholar] [CrossRef]
- Dugo, L.; Collin, M.; Thiemermann, C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock 2007, 27, 113–123. [Google Scholar] [CrossRef]
- Seino, K.; Kayagaki, N.; Takeda, K.; Fukao, K.; Okumura, K.; Yagita, H. Contribution of Fas ligand to T cell-mediated hepatic injury in mice. Gastroenterology 1997, 113, 1315–1322. [Google Scholar] [CrossRef]
- Morita, M.; Watanabe, Y.; Akaike, T. Protective effect of hepatocyte growth factor on interferon-gamma-induced cytotoxicity in mouse hepatocytes. Hepatology 1995, 21, 1585–1593. [Google Scholar]
- Kano, A.; Haruyama, T.; Akaike, T.; Watanabe, Y. IRF-1 is an essential mediator in IFN-gamma-induced cell cycle arrest and apoptosis of primary cultured hepatocytes. Biochem. Biophys. Res. Commun. 1999, 257, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Kano, A.; Watanabe, Y.; Takeda, N.; Aizawa, S.; Akaike, T. Analysis of IFN-gamma-induced cell cycle arrest and cell death in hepatocytes. J. Biochem. 1997, 121, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Horras, C.J.; Lamb, C.L.; Mitchell, K.A. Regulation of hepatocyte fate by interferon-gamma. Cytokine Growth Factor Rev. 2011, 22, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodovotz, Y.; Kim, P.K.; Bagci, E.Z.; Ermentrout, G.B.; Chow, C.C.; Bahar, I.; Billiar, T.R. Inflammatory modulation of hepatocyte apoptosis by nitric oxide: In vivo, in vitro, and in silico studies. Curr. Mol. Med. 2004, 4, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Oh, Y.K.; Rhee, M.; Lim, J.Y.; Hwang, J.Y.; Park, Y.S.; Kwon, Y.; Choi, K.H.; Jo, I.; Park, S.I.; et al. The role of STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial transmembrane potential during hepatic cell death induced by LPS/d-GalN. J. Mol. Biol. 2007, 369, 967–984. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Brenner, D.A. Role of glycogen synthase kinase-3 in TNF-alpha-induced NF-kappaB activation and apoptosis in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G204–G211. [Google Scholar] [CrossRef]
- Hatano, E.; Bennett, B.L.; Manning, A.M.; Qian, T.; Lemasters, J.J.; Brenner, D.A. NF-kappaB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis. Gastroenterology 2001, 120, 1251–1262. [Google Scholar] [CrossRef]
- Kandar, C.C.; Sen, D.; Maity, A. Anti-inflammatory potential of GSK-3 inhibitors. Curr. Drug Targets 2021, 22, 1464–1476. [Google Scholar] [CrossRef]
- Cortes-Vieyra, R.; Silva-Garcia, O.; Gomez-Garcia, A.; Gutierrez-Castellanos, S.; Alvarez-Aguilar, C.; Baizabal-Aguirre, V.M. Glycogen synthase kinase 3 beta modulates the inflammatory response activated by bacteria, viruses, and parasites. Front. Immunol. 2021, 12, 675751. [Google Scholar] [CrossRef]
- Roca, C.; Campillo, N.E. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2016–2019). Expert Opin. Ther. Patents 2020, 30, 863–872. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hepatic Injury Model | The Blockade of GSK-3 | References |
|---|---|---|
| Zymosan | 4-Benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8) | [35] |
| IRI | SB216763/TDZD-8/Carbon monoxide | [36,37,48] |
| Carbon tetrachloride | Methane | [39] |
| LPS/D-GalN | 4-Phenylbutyric acid/SB216763 | [43,44,49,50] |
| LPS | Lithium chloride (LiCl) | [45] |
| CLP | SB216763 | [46] |
| Hemorrhagic shock | TDZD-8 | [47] |
| Transplantation | Suberoylanilide hydroxamic acid | [51] |
| Lead | Curcumin/l-carnitine | [52,53] |
| HCV | LiCl | [54] |
| Palmitate | GSK-3 inhibitor IX/Enzastaurin | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-L.; Tseng, P.-C.; Satria, R.D.; Nguyen, T.T.; Tsai, C.-C.; Lin, C.-F. Role of Glycogen Synthase Kinase-3 in Interferon-γ-Mediated Immune Hepatitis. Int. J. Mol. Sci. 2022, 23, 4669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094669
Chen C-L, Tseng P-C, Satria RD, Nguyen TT, Tsai C-C, Lin C-F. Role of Glycogen Synthase Kinase-3 in Interferon-γ-Mediated Immune Hepatitis. International Journal of Molecular Sciences. 2022; 23(9):4669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094669
Chicago/Turabian StyleChen, Chia-Ling, Po-Chun Tseng, Rahmat Dani Satria, Thi Thuy Nguyen, Cheng-Chieh Tsai, and Chiou-Feng Lin. 2022. "Role of Glycogen Synthase Kinase-3 in Interferon-γ-Mediated Immune Hepatitis" International Journal of Molecular Sciences 23, no. 9: 4669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094669