
NAD+ Precursors Repair Mitochondrial Function in Diabetes and Prevent Experimental Diabetic Neuropathy

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Administration of NMN Prevents STZ-Induced Increases in Triglyceride and Non-Esterified Fatty Lipid Levels in Rat Blood
2.2. Administration of NMN Prevents STZ-Induced Peripheral Neuropathy in Rats
2.3. Administration of NMN Prevents STZ-Induced Peripheral Neuropathy in Mice
2.4. Dietary Administration of NR Prevents HFD-Induced Increases in Triglyceride and NEFA Levels in Mice
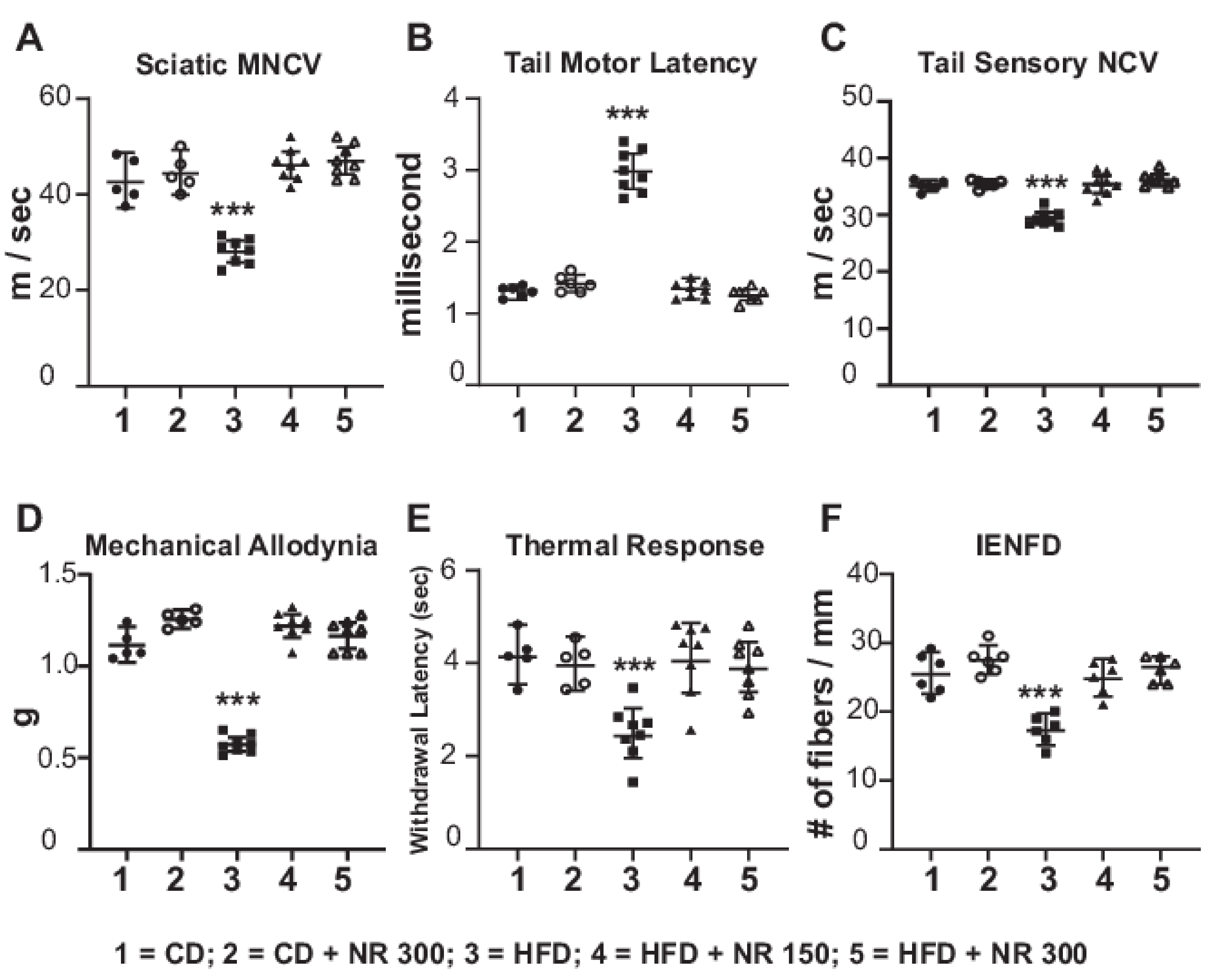
2.5. Dietary Administration of NR Prevents Peripheral Neuropathy Induced by an HFD
2.6. Administration of NMN or NR Corrects STZ- or HFD-Induced Alterations in the NAD+ Metabolome
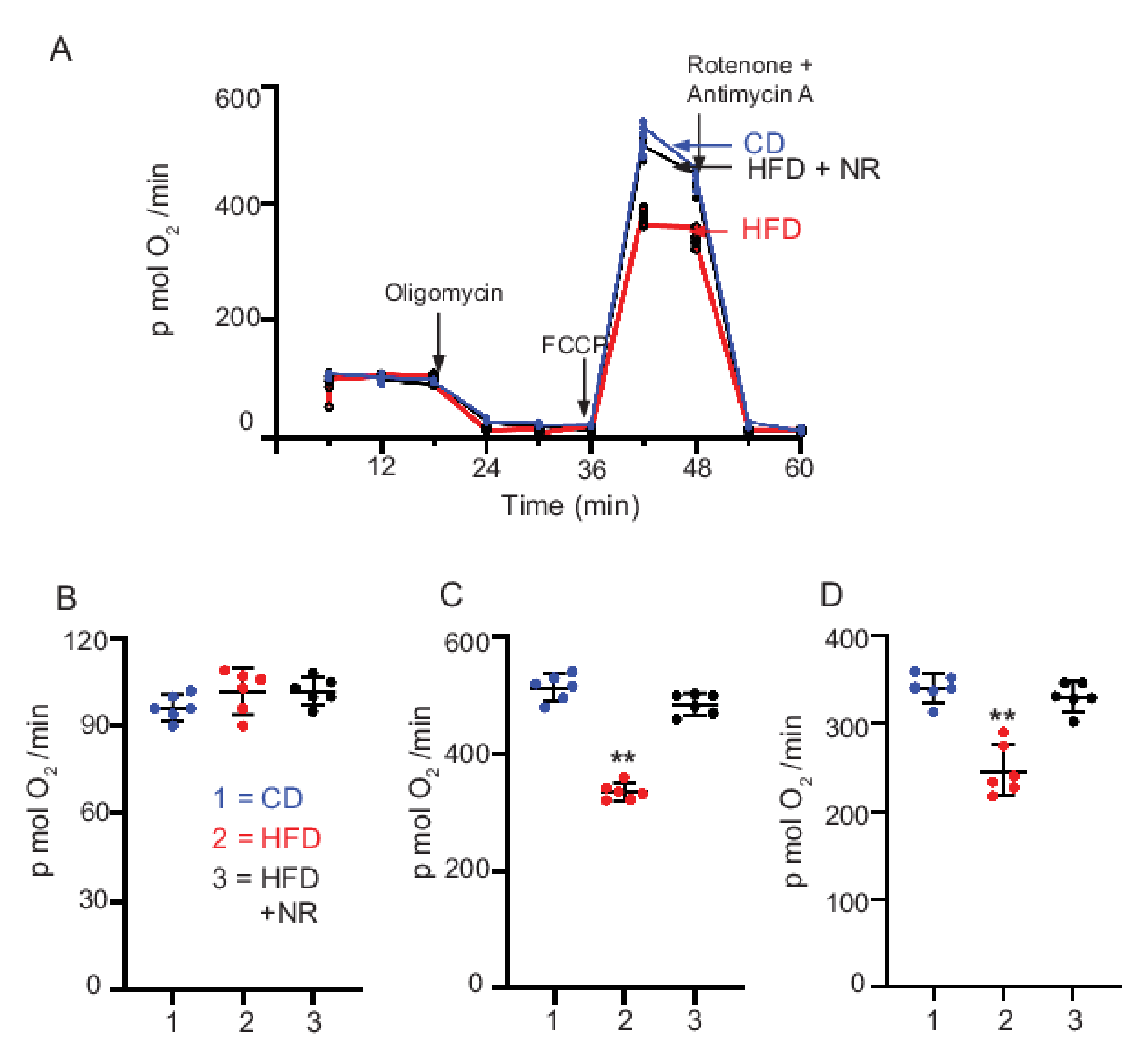
2.7. Dietary Supplementation with NR Maintains Mt Bioenergetic Function
3. Discussion
3.1. Diabetes and HFD Alter NAD+ Metabolic Pathways
3.2. Preservation of NAD+ Rescues Axonal Degeneration
3.3. NAD+ Precursors Protect Alterations in Metabolic Syndrome
3.4. HFD-Induced Decrease in Mt Respiration Capacity Was Rescued by NR Administration
4. Materials and Methods
4.1. Diabetes Induction with STZ
4.2. T2D Diabetes Induction with HFD
4.3. Experimental Design
4.4. Neuropathy Measurements
4.5. Mt Respiration
4.6. Quantification of NAD+
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Zilliox, L.A.; Russell, J.W. Physical activity and dietary interventions in diabetic neuropathy: A systematic review. Clin. Auton. Res. 2019, 29, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Rumora, A.E.; Lentz, S.I.; Hinder, L.M.; Jackson, S.W.; Valesano, A.; Levinson, G.E.; Feldman, E.L. Dyslipidemia impairs mitochondrial trafficking and function in sensory neurons. FASEB J. 2018, 32, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop-Busui, R.; Boulton, A.J.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic Neuropathy: A Position Statement by the American Diabetes Association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Eid, S.; Sas, K.M.; Abcouwer, S.F.; Feldman, E.L.; Gardner, T.W.; Pennathur, S.; Fort, P.E. New insights into the mechanisms of diabetic complications: Role of lipids and lipid metabolism. Diabetologia 2019, 62, 1539–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viader, A.; Golden, J.P.; Baloh, R.H.; Schmidt, R.E.; Hunter, D.A.; Milbrandt, J. Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. J. Neurosci. 2011, 31, 10128–10140. [Google Scholar] [CrossRef]
- Viader, A.; Sasaki, Y.; Kim, S.; Strickland, A.; Workman, C.S.; Yang, K.; Gross, R.W.; Milbrandt, J. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 2013, 77, 886–898. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Ravipati, A.; Nimmagadda, V.; Schubert, M.; Castellani, R.J.; Russell, J.W. Potential roles of PINK1 for increased PGC-1alpha-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion 2014, 18, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Feldman, E.L.; Nave, K.A.; Jensen, T.S.; Bennett, D.L.H. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef] [Green Version]
- Rumora, A.E.; Savelieff, M.G.; Sakowski, S.A.; Feldman, E.L. Disorders of mitochondrial dynamics in peripheral neuropathy: Clues from hereditary neuropathy and diabetes. Int. Rev. Neurobiol. 2019, 145, 127–176. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Salimian, M.; Konduru, S.R.; Choi, J.; Kumar, P.; Long, A.; Klimova, N.; Ho, C.Y.; Kristian, T.; Russell, J.W. Overexpression of Sirtuin 1 protein in neurons prevents and reverses experimental diabetic neuropathy. Brain 2019, 142, 3737–3752. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, K.; Anjaneyulu, M.; Choi, J.; Kumar, P.; Salimian, M.; Ho, C.Y.; Russell, J.W. Role of mitochondria in diabetic peripheral neuropathy: Influencing the NAD(+)-dependent SIRT1-PGC-1alpha-TFAM pathway. Int. Rev. Neurobiol. 2019, 145, 177–209. [Google Scholar] [CrossRef] [PubMed]
- Demare, S.; Kothari, A.; Calcutt, N.A.; Fernyhough, P. Metformin as a potential therapeutic for neurological disease: Mobilizing AMPK to repair the nervous system. Expert Rev. Neurother. 2021, 21, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Schartner, E.; Sabbir, M.G.; Saleh, A.; Silva, R.V.; Roy Chowdhury, S.; Smith, D.R.; Fernyhough, P. High glucose concentration suppresses a SIRT2 regulated pathway that enhances neurite outgrowth in cultured adult sensory neurons. Exp. Neurol. 2018, 309, 134–147. [Google Scholar] [CrossRef]
- Sasaki, Y. Metabolic aspects of neuronal degeneration: From a NAD(+) point of view. Neurosci. Res. 2019, 139, 9–20. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Choi, J.; Arvas, M.I.; Salimian, M.; Singh, S.; Xu, S.; Gullapalli, R.P.; Kristian, T.; Russell, J.W. Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons. Int. J. Mol. Sci. 2020, 21, 3756. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Trammell, S.A.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef] [Green Version]
- Gerdts, J.; Summers, D.W.; Milbrandt, J.; DiAntonio, A. Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 2016, 89, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Coleman, M.P.; Hoke, A. Programmed axon degeneration: From mouse to mechanism to medicine. Nat. Rev. Neurosci. 2020, 21, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Doan, R.A.; Strickland, A.; Huang, X.; Milbrandt, J.; DiAntonio, A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139, 3092–3108. [Google Scholar] [CrossRef] [Green Version]
- Turkiew, E.; Falconer, D.; Reed, N.; Hoke, A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 2017, 22, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Xu, W.; Lyzogubov, V.V.; Ilnytska, O.; Mashtalir, N.; Vareniuk, I.; Pavlov, I.A.; Zhang, J.; Slusher, B.; Drel, V.R. PARP inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy. Free Radic. Biol. Med. 2008, 44, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Escande, C.; Chini, C.C.; Nin, V.; Dykhouse, K.M.; Novak, C.M.; Levine, J.; van Deursen, J.; Gores, G.J.; Chen, J.; Lou, Z.; et al. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J. Clin. Investig. 2010, 120, 545–558. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Araki, T.; Milbrandt, J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J. Neurosci. 2006, 26, 8484–8491. [Google Scholar] [CrossRef] [Green Version]
- Press, C.; Milbrandt, J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J. Neurosci. 2008, 28, 4861–4871. [Google Scholar] [CrossRef] [Green Version]
- Yahata, N.; Yuasa, S.; Araki, T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J. Neurosci. 2009, 29, 6276–6284. [Google Scholar] [CrossRef]
- Babetto, E.; Beirowski, B.; Janeckova, L.; Brown, R.; Gilley, J.; Thomson, D.; Ribchester, R.R.; Coleman, M.P. Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo. J. Neurosci. 2010, 30, 13291–13304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, M.; Loreto, A.; Orsomando, G.; Mori, V.; Zamporlini, F.; Hulse, R.P.; Webster, J.; Donaldson, L.F.; Gering, M.; Raffaelli, N.; et al. NMN Deamidase Delays Wallerian Degeneration and Rescues Axonal Defects Caused by NMNAT2 Deficiency In Vivo. Curr. Biol. 2017, 27, 784–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilnytska, O.; Lyzogubov, V.V.; Stevens, M.J.; Drel, V.R.; Mashtalir, N.; Pacher, P.; Yorek, M.A.; Obrosova, I.G. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes 2006, 55, 1686–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchiroud, L.; Houtkooper, R.H.; Auwerx, J. NAD(+) metabolism: A therapeutic target for age-related metabolic disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Drel, V.R.; Szabo, C.; Stevens, M.J.; Obrosova, I.G. Low-dose poly(ADP-ribose) polymerase inhibitor-containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes 2005, 54, 1514–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Szabo, C.; Pacher, P.; Southan, G.J.; Abatan, O.I.; Charniauskaya, T.; Stevens, M.J.; Obrosova, I.G. Evaluation of orally active poly(ADP-ribose) polymerase inhibitor in streptozotocin-diabetic rat model of early peripheral neuropathy. Diabetologia 2004, 47, 710–717. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Liu, J.; Luan, Y.; Liu, Z.; Lai, H.; Zhong, W.; Yang, Y.; Yu, H.; Feng, N.; Wang, H.; et al. Sarm1 Gene Deficiency Attenuates Diabetic Peripheral Neuropathy in Mice. Diabetes 2019, 68, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 26–33. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilley, J.; Coleman, M.P. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010, 8, e1000300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilley, J.; Ribchester, R.R.; Coleman, M.P. Sarm1 Deletion, but Not Wld(S), Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy. Cell Rep. 2017, 21, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Vohra, B.P.; Baloh, R.H.; Milbrandt, J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 2009, 29, 6526–6534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gille, A.; Bodor, E.T.; Ahmed, K.; Offermanns, S. Nicotinic acid: Pharmacological effects and mechanisms of action. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 79–106. [Google Scholar] [CrossRef]
- Conze, D.; Brenner, C.; Kruger, C.L. Safety and Metabolism of Long-term Administration of NIAGEN (Nicotinamide Riboside Chloride) in a Randomized, Double-Blind, Placebo-controlled Clinical Trial of Healthy Overweight Adults. Sci. Rep. 2019, 9, 9772. [Google Scholar] [CrossRef]
- Irie, J.; Inagaki, E.; Fujita, M.; Nakaya, H.; Mitsuishi, M.; Yamaguchi, S.; Yamashita, K.; Shigaki, S.; Ono, T.; Yukioka, H.; et al. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr. J. 2020, 67, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.J.; Li, F.; Drel, V.R.; Abatan, O.I.; Kim, H.; Burnett, D.; Larkin, D.; Obrosova, I.G. Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J. Pharmacol. Exp. Ther. 2007, 320, 458–464. [Google Scholar] [CrossRef] [Green Version]
- Knip, M.; Douek, I.F.; Moore, W.P.; Gillmor, H.A.; McLean, A.E.; Bingley, P.J.; Gale, E.A.; European Nicotinamide Diabetes Intervention Trial Group. Safety of high-dose nicotinamide: A review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Ramsden, D. Nicotinamide: A double edged sword. Parkinsonism Relat. Disord. 2005, 11, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.G.; Zhao, G.; Qin, Q.; Wang, B.; Liu, L.; Liu, Y.; Deng, S.C.; Tian, K.; Wang, C.Y. Nicotinamide prohibits proliferation and enhances chemosensitivity of pancreatic cancer cells through deregulating SIRT1 and Ras/Akt pathways. Pancreatology 2013, 13, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Peled, T.; Shoham, H.; Aschengrau, D.; Yackoubov, D.; Frei, G.; Rosenheimer, G.N.; Lerrer, B.; Cohen, H.Y.; Nagler, A.; Fibach, E.; et al. Nicotinamide, a SIRT1 inhibitor, inhibits differentiation and facilitates expansion of hematopoietic progenitor cells with enhanced bone marrow homing and engraftment. Exp. Hematol. 2012, 40, 342–355.e341. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Trammell, S.A.; Brenner, C. NNMT: A Bad Actor in Fat Makes Good in Liver. Cell Metab. 2015, 22, 200–201. [Google Scholar] [CrossRef] [Green Version]
- Ruf, S.; Hallur, M.S.; Anchan, N.K.; Swamy, I.N.; Murugesan, K.R.; Sarkar, S.; Narasimhulu, L.K.; Putta, V.; Shaik, S.; Chandrasekar, D.V.; et al. Novel nicotinamide analog as inhibitor of nicotinamide N-methyltransferase. Bioorg. Med. Chem. Lett. 2018, 28, 922–925. [Google Scholar] [CrossRef]
- Marshall, A.; Alam, U.; Themistocleous, A.; Calcutt, N.; Marshall, A. Novel and Emerging Electrophysiological Biomarkers of Diabetic Neuropathy and Painful Diabetic Neuropathy. Clin. Ther. 2021, 43, 1441–1456. [Google Scholar] [CrossRef]
- Drexel, H. Nicotinic acid in the treatment of hyperlipidaemia. Fundam. Clin. Pharmacol. 2007, 21, 5–6. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Imamura, H.; Nagayama, D.; Ishihara, N.; Tanaka, S.; Watanabe, R.; Watanabe, Y.; Sato, Y.; Yamaguchi, T.; Ban, N.; Kawana, H.; et al. Resveratrol attenuates triglyceride accumulation associated with upregulation of Sirt1 and lipoprotein lipase in 3T3-L1 adipocytes. Mol. Genet. Metab. Rep. 2017, 12, 44–50. [Google Scholar] [CrossRef]
- Fernyhough, P.; Calcutt, N.A. New Directions in Diabetic Neuropathy: Evolution or Extinction? Int. Rev. Neurobiol. 2016, 127, 229–234. [Google Scholar] [CrossRef]
- Benyo, Z.; Gille, A.; Kero, J.; Csiky, M.; Suchankova, M.C.; Nusing, R.M.; Moers, A.; Pfeffer, K.; Offermanns, S. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J. Clin. Investig. 2005, 115, 3634–3640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunaru, S.; Kero, J.; Schaub, A.; Wufka, C.; Blaukat, A.; Pfeffer, K.; Offermanns, S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med. 2003, 9, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [Green Version]
- Sauve, A.A.; Wolberger, C.; Schramm, V.L.; Boeke, J.D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006, 75, 435–465. [Google Scholar] [CrossRef]
- Roy Chowdhury, S.K.; Smith, D.R.; Saleh, A.; Schapansky, J.; Marquez, A.; Gomes, S.; Akude, E.; Morrow, D.; Calcutt, N.A.; Fernyhough, P. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 2012, 135, 1751–1766. [Google Scholar] [CrossRef]
- Sadi, G.; Konat, D. Resveratrol regulates oxidative biomarkers and antioxidant enzymes in the brain of streptozotocin-induced diabetic rats. Pharm. Biol. 2016, 54, 1156–1163. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lian, S.; Zhang, J.; Lin, D.; Huang, C.; Liu, L.; Chen, Z. Mitochondrial Perturbation Contributing to Cognitive Decline in Streptozotocin-Induced Type 1 Diabetic Rats. Cell Physiol. Biochem. 2018, 46, 1668–1682. [Google Scholar] [CrossRef]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xu, S.; Lin, L.; Gu, X.; Fu, C.; Fang, Y.; Li, X.; Wang, X. High-fat Diet Mediates Anxiolytic-like Behaviors in a Time-dependent Manner Through the Regulation of SIRT1 in the Brain. Neuroscience 2018, 372, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Bril, V.; Calcutt, N.A.; Cameron, N.E.; Cotter, M.A.; Dobrowsky, R.; Feldman, E.L.; Fernyhough, P.; Jakobsen, J.; Malik, R.A.; et al. Phenotyping animal models of diabetic neuropathy: A consensus statement of the diabetic neuropathy study group of the EASD (Neurodiab). J. Peripher. Nerv. Syst. 2014, 19, 77–87. [Google Scholar] [CrossRef]
- Choi, J.; Chandrasekaran, K.; Demarest, T.G.; Kristian, T.; Xu, S.; Vijaykumar, K.; Dsouza, K.G.; Qi, N.R.; Yarowsky, P.J.; Gallipoli, R.; et al. Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Ann. Clin. Transl. Neurol. 2014, 1, 589–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, K.; Muragundla, A.; Demarest, T.G.; Choi, J.; Sagi, A.R.; Najimi, N.; Kumar, P.; Singh, A.; Ho, C.Y.; Fiskum, G.; et al. mGluR2/3 activation of the SIRT1 axis preserves mitochondrial function in diabetic neuropathy. Ann. Clin. Transl. Neurol. 2017, 4, 844–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauria, G.; Hsieh, S.T.; Johansson, O.; Kennedy, W.R.; Leger, J.M.; Mellgren, S.I.; Nolano, M.; Merkies, I.S.; Polydefkis, M.; Smith, A.G.; et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur. J. Neurol. 2010, 17, 903–912, e944–909. [Google Scholar] [CrossRef]
- Lauria, G.; Lombardi, R.; Borgna, M.; Penza, P.; Bianchi, R.; Savino, C.; Canta, A.; Nicolini, G.; Marmiroli, P.; Cavaletti, G. Intraepidermal nerve fiber density in rat foot pad: Neuropathologic-neurophysiologic correlation. J. Peripher. Nerv. Syst. 2005, 10, 202–208. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Anjaneyulu, M.; Inoue, T.; Choi, J.; Sagi, A.R.; Chen, C.; Ide, T.; Russell, J.W. Mitochondrial transcription factor A regulation of mitochondrial degeneration in experimental diabetic neuropathy. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E132–E141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Yang, L.; Qin, A.R.; Ly, J.; Liederer, B.M.; Messick, K.; Ma, S.; Zak, M.; Dragovich, P.S.; Dean, B.J.; et al. Measuring NAD(+) levels in mouse blood and tissue samples via a surrogate matrix approach using LC-MS/MS. Bioanalysis 2014, 6, 1445–1457. [Google Scholar] [CrossRef]
- Russell, J.W.; Berent-Spillson, A.; Vincent, A.M.; Freimann, C.L.; Sullivan, K.A.; Feldman, E.L. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol. Dis. 2008, 30, 420–429. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Parameters | WT | Significance | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Non-Dia (n = 6) | Non-Dia + NMN (100 mg/kg) (n = 6) | STZ (n = 6) | STZ + NMN (50 mg/kg) (n = 6) | STZ + NMN (100 mg /kg) (n = 6) | 1 vs. 2 | 1 vs. 3 | 3 vs. 4 | 3 vs. 5 | |
| Group # | 1 | 2 | 3 | 4 | 5 | ||||
| Body Weight (g) | 450 ± 17 | 431 ± 19 | 346 ± 14 | 345 ± 13 | 331 ± 14 | NS | <0.001 | NS | NS |
| Plasma Glucose (mg/dL) | 132 ± 18 | 128 ± 17 | 405 ± 22 | 399 ± 13 | 388 ± 15 | NS | <0.001 | NS | NS |
| HbA1c% | 5.4 ± 1 | 5.4 ± 1 | 16.7 ± 4.1 | 15.8 ± 2.6 | 15.3 ± 2.9 | NS | <0.01 | NS | NS |
| Insulin (mµ/mL) | 8.3 ± 2 | 8 ± 2 | 1.2 ± 0.7 | 1.3 ± 0.4 | 1.6 ± 0.3 | NS | <0.001 | NS | NS |
| Total Cholesterol (mg/dL) | 68 ± 13 | 59 ± 11 | 173 ± 14 | 133 ± 71 | 139 ± 48 | NS | <0.01 | NS | NS |
| Triglycerides (mg/dL) | 94 ± 11 | 96 ± 9 | 493 ± 23 | 376 ± 22 | 361 ± 19 | NS | <0.001 | <0.05 | <0.05 |
| HDL (mg/dL) | 79 ± 4 | 78 ± 5 | 128 ± 21 | 119 ± 15 | 115 ± 16 | NS | <0.001 | NS | NS |
| LDL (mg/dL) | 13 ± 5 | 10 ± 4 | 63 ± 5 | 64 ± 9 | 60 ± 9 | NS | <0.01 | NS | NS |
| NEFA (mM) | 2.5 ± 0.8 | 3.1 ± 0.4 | 6.6 ± 1.8 | 3.6 ± 1 | 3.8 ± 1 | NS | <0.001 | <0.05 | <0.05 |
| Parameters | WT | Significance | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Non-Dia (n = 6) | Non-Dia + NMN (100 mg/kg) (n =6) | STZ (n = 6) | STZ + NMN (50 mg/kg) (n = 6) | STZ + NMN (100 mg /kg) (n = 6) | 1 vs. 2 | 1 vs. 3 | 3 vs. 4 | 3 vs. 5 | |
| Group # | 1 | 2 | 3 | 4 | 5 | ||||
| Body Weight (g) | 30 ± 2 | 31 ± 3 | 26 ± 4 | 25 ± 3 | 21 ± 4 | NS | <0.01 | NS | NS |
| Plasma Glucose (mg/dL) | 105 ± 12 | 108 ± 11 | 405 ± 32 | 385 ± 33 | 388 ± 35 | NS | <0.001 | NS | NS |
| HbA1c% | 5.4 ± 1 | 5.4 ± 1 | 15.7 ± 3.1 | 15.8 ± 3.6 | 15.3 ± 3.4 | NS | <0.001 | NS | NS |
| Insulin (µg/L) | 0.8 ± 0.2 | 0.8 ± 0.2 | 0.2 ± 0.05 | 0.18 ± 0.04 | 0.16 ± 0.03 | NS | <0.001 | NS | NS |
| Total Cholesterol (mg/dL) | 78 ± 13 | 75 ± 21 | 173 ± 42 | 163 ± 21 | 169 ± 28 | NS | <0.01 | NS | NS |
| Triglycerides (mg/dL) | 44 ± 8 | 36 ± 9 | 93 ± 13 | 66 ± 10 | 61 ± 10 | NS | <0.001 | <0.05 | <0.05 |
| HDL (mg/dL) | 79 ± 4 | 78 ± 5 | 108 ± 21 | 99 ± 15 | 95 ± 16 | NS | <0.01 | NS | NS |
| LDL (mg/dL) | 43 ± 5 | 50 ± 4 | 83 ± 5 | 84 ± 9 | 90 ± 9 | NS | <0.01 | NS | NS |
| NEFA (mM) | 2.5 ± 0.8 | 3.1 ± 0.4 | 6.4 ± 1.4 | 3.6 ± 1 | 3.6 ± 0.8 | NS | <0.001 | <0.05 | <0.05 |
| Sciatic MNCV (m/s) | 55 ± 7 | 53 ± 6 | 26 ± 6 | 42.2 ± 7 | 46 ± 6.5 | NS | <0.001 | <0.001 | <0.001 |
| Tail MNCV (m/s) | 45 ± 4 | 43 ± 6 | 24 ± 3 | 46 ± 4.5 | 48 ± 5.3 | NS | <0.001 | <0.001 | <0.001 |
| Tail SNCV (m/s) | 41 ± 3 | 43 ± 3 | 27 ± 5 | 46 ± 7 | 48 ± 6 | NS | <0.001 | <0.001 | <0.001 |
| Von Frey Mechanical Allodynia (g) | 1.2 ± 0.2 | 1.3 ± 0.3 | 0.6 ± 0.1 | 1.0 ± 0.2 | 1.2 ± 0.3 | NS | <0.001 | <0.01 | <0.001 |
| Hargreaves Thermal Response (sec) | 12 ± 2 | 14 ± 3 | 22 ± 4 | 14 ± 3 | 12 ± 3 | NS | <0.001 | <0.001 | <0.001 |
| IENFD (# / mm) | 25 ± 3 | 23 ± 4 | 12 ± 2 | 23 ± 3 | 24 ± 3 | NS | <0.001 | <0.001 | <0.001 |
| Parameters | WT | Significance | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CD (n = 8) | CD + NR (300 mg/kg) (n = 8) | HFD (n = 8) | HFD + NR (150 mg/kg) (n = 8) | HFD + NR (300 mg/kg) (n = 8) | 1 vs. 2 | 1 vs. 3 | 3 vs. 4 | 3 vs. 5 | |
| Group # | 1 | 2 | 3 | 4 | 5 | ||||
| Body Weight (g) | 30 ± 2 | 31 ± 3 | 46 ± 4 | 45 ± 3 | 41 ± 4 | NS | <0.001 | NS | NS |
| Plasma Glucose (mg/dL) | 135 ± 18 | 148 ± 17 | 205 ± 22 | 185 ± 13 | 188 ± 15 | NS | <0.001 | NS | NS |
| HbA1c% | 5.4 ± 1 | 5.4 ± 1 | 6.7 ± 1.1 | 5.3 ± 0.6 | 5.8 ± 0.9 | NS | <0.001 | <0.001 | <0.01 |
| Insulin (µg/L) | 0.8 ± 0.2 | 0.8 ± 0.2 | 4 ± 0.5 | 3.8± 0.4 | 3.6 ± 0.3 | NS | <0.001 | NS | NS |
| Total Cholesterol (mg/dL) | 152 ± 32 | 115 ± 12 | 273 ± 42 | 233 ± 51 | 249 ± 21 | NS | <0.001 | <0.05 | <0.05 |
| Triglycerides (mg/dL) | 144 ± 18 | 136 ± 16 | 241 ± 32 | 142 ± 28 | 96 ± 16 | NS | <0.001 | <0.01 | <0.001 |
| HDL Cholesterol (mg/dL) | 142 ± 14 | 130 ± 14 | 256 ± 29 | 169 ± 15 | 203 ± 26 | NS | <0.001 | <0.001 | <0.05 |
| Non-HDL Cholesterol (mg/dL) | 10 ± 3 | 12 ± 3 | 15 ± 6 | 14 ± 2 | 10 ± 3 | NS | NS | NS | NS |
| NEFA (mM) | 3 ± 0.29 | 3.1 ± 0.4 | 5.9 ± 1 | 3.6 ± 0.7 | 4.8 ± 1 | NS | <0.001 | <0.001 | <0.05 |
| GTT-AUC (mg*min/dL X 103) | 3 ± 0.29 | 3.6 ± 0.46 | 6.1 ± 1 | 6.2 ± 1 | 5.9 ± 1.2 | NS | <0.001 | NS | NS |
| Parameters. (p mol/mg Protein) | WT | Significance | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CD (n = 11) | CD + NR (300 mg/kg) (n = 11) | HFD (n = 11) | HFD + NR (150 mg/kg) (n = 11) | HFD + NR (300 mg/kg) (n = 11) | 1 vs. 2 | 1 vs. 3 | 3 vs. 4 | 3 vs. 5 | |
| Group # | 1 | 2 | 3 | 4 | 5 | ||||
| NAD+ | 1830 ± 94 | 2010 ± 98 | 1390 ± 81 | 1880 ± 72 | 2140 ± 79 | NS | <0.001 | <0.05 | <0.01 |
| NMN | 3.8 ± 0.6 | 4.4 ± 0.7 | 4.1 ± 0.8 | 4.6 ± 0.9 | 4.7 ± 0.9 | NS | NS | NS | NS |
| NR | 3.8 ± 0.7 | 5.9 ± 0.8 | 3.6 ± 0.8 | 5.65 ± 0.5 | 5.6 ± 0.6 | <0.05 | NS | <0.05 | <0.05 |
| Nam | 130 ± 13 | 270 ± 14 | 190 ± 24 | 200 ± 23 | 210 ± 26 | <0.005 | <0.05 | NS | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekaran, K.; Najimi, N.; Sagi, A.R.; Yarlagadda, S.; Salimian, M.; Arvas, M.I.; Hedayat, A.F.; Kevas, Y.; Kadakia, A.; Russell, J.W. NAD+ Precursors Repair Mitochondrial Function in Diabetes and Prevent Experimental Diabetic Neuropathy. Int. J. Mol. Sci. 2022, 23, 4887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094887
Chandrasekaran K, Najimi N, Sagi AR, Yarlagadda S, Salimian M, Arvas MI, Hedayat AF, Kevas Y, Kadakia A, Russell JW. NAD+ Precursors Repair Mitochondrial Function in Diabetes and Prevent Experimental Diabetic Neuropathy. International Journal of Molecular Sciences. 2022; 23(9):4887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094887
Chicago/Turabian StyleChandrasekaran, Krish, Neda Najimi, Avinash R. Sagi, Sushuma Yarlagadda, Mohammad Salimian, Muhammed Ikbal Arvas, Ahmad F. Hedayat, Yanni Kevas, Anand Kadakia, and James W. Russell. 2022. "NAD+ Precursors Repair Mitochondrial Function in Diabetes and Prevent Experimental Diabetic Neuropathy" International Journal of Molecular Sciences 23, no. 9: 4887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094887